Optic Atrophy-Associated TMEM126A Is an Assembly Factor for the ND4- Module of Mitochondrial Complex I
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

NDUFAF1 Antibody
Efficient Professional Protein and Antibody Platforms NDUFAF1 Antibody Basic information: Catalog No.: UPA63763 Source: Rabbit Size: 50ul/100ul Clonality: monoclonal Concentration: 1mg/ml Isotype: Rabbit IgG Purification: Protein A purified. Useful Information: WB:1:1000 ICC:1:50-1:200 Applications: IHC:1:50-1:200 FC:1:50-1:100 Reactivity: Human Specificity: This antibody recognizes NDUFAF1 protein. Immunogen: Synthetic peptide within C terminal human NDUFAF1. This gene encodes a complex I assembly factor protein. Complex I (NADH-ubiquinone oxidoreductase) catalyzes the transfer of electrons from NADH to ubiquinone (coenzyme Q) in the first step of the mitochondrial respiratory chain, resulting in the translocation of protons across the inner mitochondrial membrane. The encoded protein is required for assembly of complex I, and mutations in this gene are a cause of mitochondrial complex I deficiency. Alternatively spliced transcript variants have been observed for Description: this gene, and a pseudogene of this gene is located on the long arm of chromosome 19. Part of the mitochondrial complex I assembly (MCIA) com- plex. The complex comprises at least TMEM126B, NDUFAF1, ECSIT, and ACAD9. Interacts with ECSIT. Interacts with ACAD9. At early stages of com- plex I assembly, it is found in intermediate subcomplexes that contain dif- ferent subunits including NDUFB6, NDUFA6, NDUFA9, NDUFS3, NDUFS7, ND1, ND2 and ND3 Uniprot: Q9Y375 Human BiowMW: 38 kDa Buffer: 1*TBS (pH7.4), 1%BSA, 50%Glycerol. Preservative: 0.05% Sodium Azide. Storage: Store at 4°C short term and -20°C long term. Avoid freeze-thaw cycles. Note: For research use only, not for use in diagnostic procedure. -

Molecular Mechanism of ACAD9 in Mitochondrial Respiratory Complex 1 Assembly
bioRxiv preprint doi: https://doi.org/10.1101/2021.01.07.425795; this version posted January 9, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Molecular mechanism of ACAD9 in mitochondrial respiratory complex 1 assembly Chuanwu Xia1, Baoying Lou1, Zhuji Fu1, Al-Walid Mohsen2, Jerry Vockley2, and Jung-Ja P. Kim1 1Department of Biochemistry, Medical College of Wisconsin, Milwaukee, Wisconsin, 53226, USA 2Department of Pediatrics, University of Pittsburgh School of Medicine, University of Pittsburgh, Children’s Hospital of Pittsburgh of UPMC, Pittsburgh, PA 15224, USA Abstract ACAD9 belongs to the acyl-CoA dehydrogenase family, which catalyzes the α-β dehydrogenation of fatty acyl-CoA thioesters. Thus, it is involved in fatty acid β-oxidation (FAO). However, it is now known that the primary function of ACAD9 is as an essential chaperone for mitochondrial respiratory complex 1 assembly. ACAD9 interacts with ECSIT and NDUFAF1, forming the mitochondrial complex 1 assembly (MCIA) complex. Although the role of MCIA in the complex 1 assembly pathway is well studied, little is known about the molecular mechanism of the interactions among these three assembly factors. Our current studies reveal that when ECSIT interacts with ACAD9, the flavoenzyme loses the FAD cofactor and consequently loses its FAO activity, demonstrating that the two roles of ACAD9 are not compatible. ACAD9 binds to the carboxy-terminal half (C-ECSIT), and NDUFAF1 binds to the amino-terminal half of ECSIT. Although the binary complex of ACAD9 with ECSIT or with C-ECSIT is unstable and aggregates easily, the ternary complex of ACAD9-ECSIT-NDUFAF1 (i.e., the MCIA complex) is soluble and extremely stable. -

An Essential Role for ECSIT in Mitochondrial Complex I Assembly and Mitophagy in Macrophages
Article An Essential Role for ECSIT in Mitochondrial Complex I Assembly and Mitophagy in Macrophages Graphical Abstract Authors Fla´ via R.G. Carneiro, Alice Lepelley, John J. Seeley, Matthew S. Hayden, Sankar Ghosh Correspondence [email protected] In Brief Macrophages rely on fine-tuning their metabolism to fulfill their anti-bacterial functions. Carneiro et al. show that the complex I assembly factor ECSIT is an essential regulator of the balance between mitochondrial respiration and glycolysis and the maintenance of a healthy mitochondrial pool through mitophagy. Highlights d Loss of ECSIT in macrophages leads to a striking glycolytic shift d ECSIT is essential for complex I assembly and stability in macrophages d Role of ECSIT in mROS production and removal of damaged mitochondria by mitophagy Carneiro et al., 2018, Cell Reports 22, 2654–2666 March 6, 2018 ª 2018 The Author(s). https://doi.org/10.1016/j.celrep.2018.02.051 Cell Reports Article An Essential Role for ECSIT in Mitochondrial Complex I Assembly and Mitophagy in Macrophages Fla´ via R.G. Carneiro,1,3,4 Alice Lepelley,1,4 John J. Seeley,1 Matthew S. Hayden,1,2 and Sankar Ghosh1,5,* 1Department of Microbiology and Immunology, Vagelos College of Physicians and Surgeons, Columbia University, New York, NY 10032, USA 2Section of Dermatology, Department of Surgery, Dartmouth-Hitchcock Medical Center, Lebanon, NH 03756, USA 3FIOCRUZ, Center for Technological Development in Health (CDTS), Rio de Janeiro, Brazil 4These authors contributed equally 5Lead Contact *Correspondence: [email protected] https://doi.org/10.1016/j.celrep.2018.02.051 SUMMARY 2015). There, ECSIT-dependent mROS production promotes activation of the phagosomal nicotinamide adenine dinucleotide ECSIT is a mitochondrial complex I (CI)-associated phosphate (NADPH) oxidase system and ROS-dependent protein that has been shown to regulate the pro- killing of engulfed microbes (West et al., 2011). -

Targeting MYCN in Neuroblastoma by BET Bromodomain Inhibition
Published OnlineFirst February 21, 2013; DOI: 10.1158/2159-8290.CD-12-0418 RESEARCH ARTICLE Targeting MYCN in Neuroblastoma by BET Bromodomain Inhibition Alexandre Puissant1,3, Stacey M. Frumm1,3, Gabriela Alexe1,3,5,6, Christopher F. Bassil1,3, Jun Qi2, Yvan H. Chanthery8, Erin A. Nekritz8, Rhamy Zeid2, William Clay Gustafson8, Patricia Greninger7, Matthew J. Garnett10, Ultan McDermott10, Cyril H. Benes7, Andrew L. Kung1,3, William A. Weiss8,9, James E. Bradner2,4, and Kimberly Stegmaier1,3,6 Downloaded from cancerdiscovery.aacrjournals.org on October 2, 2021. © 2013 American Association for Cancer Research. 15-CD-12-0418_p308-323.indd 1 22/02/13 12:15 AM Published OnlineFirst February 21, 2013; DOI: 10.1158/2159-8290.CD-12-0418 A BSTRACT Bromodomain inhibition comprises a promising therapeutic strategy in cancer, particularly for hematologic malignancies. To date, however, genomic biomarkers to direct clinical translation have been lacking. We conducted a cell-based screen of genetically defined cancer cell lines using a prototypical inhibitor of BET bromodomains. Integration of genetic features with chemosensitivity data revealed a robust correlation between MYCN amplification and sensitivity to bromodomain inhibition. We characterized the mechanistic and translational significance of this finding in neuroblastoma, a childhood cancer with frequent amplification of MYCN. Genome-wide expression analysis showed downregulation of the MYCN transcriptional program accompanied by suppression of MYCN transcription. Functionally, bromodomain-mediated inhibition of MYCN impaired growth and induced apoptosis in neuroblastoma. BRD4 knockdown phenocopied these effects, establishing BET bromodomains as transcriptional regulators of MYCN. BET inhibition conferred a significant survival advantage in 3 in vivo neuroblastoma models, providing a compelling rationale for developing BET bro- modomain inhibitors in patients with neuroblastoma. -

Mitoxplorer, a Visual Data Mining Platform To
mitoXplorer, a visual data mining platform to systematically analyze and visualize mitochondrial expression dynamics and mutations Annie Yim, Prasanna Koti, Adrien Bonnard, Fabio Marchiano, Milena Dürrbaum, Cecilia Garcia-Perez, José Villaveces, Salma Gamal, Giovanni Cardone, Fabiana Perocchi, et al. To cite this version: Annie Yim, Prasanna Koti, Adrien Bonnard, Fabio Marchiano, Milena Dürrbaum, et al.. mitoXplorer, a visual data mining platform to systematically analyze and visualize mitochondrial expression dy- namics and mutations. Nucleic Acids Research, Oxford University Press, 2020, 10.1093/nar/gkz1128. hal-02394433 HAL Id: hal-02394433 https://hal-amu.archives-ouvertes.fr/hal-02394433 Submitted on 4 Dec 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License Nucleic Acids Research, 2019 1 doi: 10.1093/nar/gkz1128 Downloaded from https://academic.oup.com/nar/advance-article-abstract/doi/10.1093/nar/gkz1128/5651332 by Bibliothèque de l'université la Méditerranée user on 04 December 2019 mitoXplorer, a visual data mining platform to systematically analyze and visualize mitochondrial expression dynamics and mutations Annie Yim1,†, Prasanna Koti1,†, Adrien Bonnard2, Fabio Marchiano3, Milena Durrbaum¨ 1, Cecilia Garcia-Perez4, Jose Villaveces1, Salma Gamal1, Giovanni Cardone1, Fabiana Perocchi4, Zuzana Storchova1,5 and Bianca H. -

New Perspective in Diagnostics of Mitochondrial Disorders
Pronicka et al. J Transl Med (2016) 14:174 DOI 10.1186/s12967-016-0930-9 Journal of Translational Medicine RESEARCH Open Access New perspective in diagnostics of mitochondrial disorders: two years’ experience with whole‑exome sequencing at a national paediatric centre Ewa Pronicka1,2*, Dorota Piekutowska‑Abramczuk1†, Elżbieta Ciara1†, Joanna Trubicka1†, Dariusz Rokicki2, Agnieszka Karkucińska‑Więckowska3, Magdalena Pajdowska4, Elżbieta Jurkiewicz5, Paulina Halat1, Joanna Kosińska6, Agnieszka Pollak7, Małgorzata Rydzanicz6, Piotr Stawinski7, Maciej Pronicki3, Małgorzata Krajewska‑Walasek1 and Rafał Płoski6* Abstract Background: Whole-exome sequencing (WES) has led to an exponential increase in identification of causative vari‑ ants in mitochondrial disorders (MD). Methods: We performed WES in 113 MD suspected patients from Polish paediatric reference centre, in whom routine testing failed to identify a molecular defect. WES was performed using TruSeqExome enrichment, followed by variant prioritization, validation by Sanger sequencing, and segregation with the disease phenotype in the family. Results: Likely causative mutations were identified in 67 (59.3 %) patients; these included variants in mtDNA (6 patients) and nDNA: X-linked (9 patients), autosomal dominant (5 patients), and autosomal recessive (47 patients, 11 homozygotes). Novel variants accounted for 50.5 % (50/99) of all detected changes. In 47 patients, changes in 31 MD-related genes (ACAD9, ADCK3, AIFM1, CLPB, COX10, DLD, EARS2, FBXL4, MTATP6, MTFMT, MTND1, MTND3, MTND5, NAXE, NDUFS6, NDUFS7, NDUFV1, OPA1, PARS2, PC, PDHA1, POLG, RARS2, RRM2B, SCO2, SERAC1, SLC19A3, SLC25A12, TAZ, TMEM126B, VARS2) were identified. The ACAD9, CLPB, FBXL4, PDHA1 genes recurred more than twice suggesting higher general/ethnic prevalence. In 19 cases, variants in 18 non-MD related genes (ADAR, CACNA1A, CDKL5, CLN3, CPS1, DMD, DYSF, GBE1, GFAP, HSD17B4, MECP2, MYBPC3, PEX5, PGAP2, PIGN, PRF1, SBDS, SCN2A) were found. -

Literature Mining Sustains and Enhances Knowledge Discovery from Omic Studies
LITERATURE MINING SUSTAINS AND ENHANCES KNOWLEDGE DISCOVERY FROM OMIC STUDIES by Rick Matthew Jordan B.S. Biology, University of Pittsburgh, 1996 M.S. Molecular Biology/Biotechnology, East Carolina University, 2001 M.S. Biomedical Informatics, University of Pittsburgh, 2005 Submitted to the Graduate Faculty of School of Medicine in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Pittsburgh 2016 UNIVERSITY OF PITTSBURGH SCHOOL OF MEDICINE This dissertation was presented by Rick Matthew Jordan It was defended on December 2, 2015 and approved by Shyam Visweswaran, M.D., Ph.D., Associate Professor Rebecca Jacobson, M.D., M.S., Professor Songjian Lu, Ph.D., Assistant Professor Dissertation Advisor: Vanathi Gopalakrishnan, Ph.D., Associate Professor ii Copyright © by Rick Matthew Jordan 2016 iii LITERATURE MINING SUSTAINS AND ENHANCES KNOWLEDGE DISCOVERY FROM OMIC STUDIES Rick Matthew Jordan, M.S. University of Pittsburgh, 2016 Genomic, proteomic and other experimentally generated data from studies of biological systems aiming to discover disease biomarkers are currently analyzed without sufficient supporting evidence from the literature due to complexities associated with automated processing. Extracting prior knowledge about markers associated with biological sample types and disease states from the literature is tedious, and little research has been performed to understand how to use this knowledge to inform the generation of classification models from ‘omic’ data. Using pathway analysis methods to better understand the underlying biology of complex diseases such as breast and lung cancers is state-of-the-art. However, the problem of how to combine literature- mining evidence with pathway analysis evidence is an open problem in biomedical informatics research. -

Understanding ACAD9 Function and the Physiologic Consequences of Its Deficiency
Title Page Understanding ACAD9 Function and the Physiologic Consequences of its Deficiency by Andrew Guelde Sinsheimer BA, Oberlin College, 2008 Submitted to the Graduate Faculty of the Department of Human Genetics Graduate School of Public Health in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Pittsburgh 2019 Committee Membership Page UNIVERSITY OF PITTSBURGH GRADUATE SCHOOL OF PUBLIC HEALTH This dissertation was presented by Andrew Guelde Sinsheimer It was defended on April 23, 2019 and approved by David Finegold, MD, Professor, Human Genetics, Graduate School of Public Health, University of Pittsburgh Eric S. Goetzman, PhD. Associate Professor, Human Genetics, Graduate School of Public Health, University of Pittsburgh Candace Kammerer, PhD, Associate Professor, Human Genetics, Graduate School of Public Health, University of Pittsburgh Dissertation Advisor: Jerry Vockley, MD, PhD, Professor, Human Genetics, Graduate School of Public Health, University of Pittsburgh ii Copyright © by Andrew Guelde Sinsheimer 2019 iii Abstract Jerry Vockley, MD, PhD Understanding ACAD9 Function and the Physiologic Consequences of its Deficiency Andrew Guelde Sinsheimer, PhD University of Pittsburgh, 2019 Abstract Acyl CoA Dehydrogenase 9 (ACAD9) is a member of the family of flavoenzymes that catalyze the dehydrogenation of Acyl-CoAs to 2,3 enoyl-CoAs in mitochondrial fatty acid oxidation (FAO). Inborn errors of metabolism of nearly all family members, including ACAD9, have been described in humans, and represent significant causes of morbidity and mortality particularly in children. ACAD9 deficiency leads to a combined defect in fatty acid oxidation and oxidative phosphorylation (OXPHOS) due to a duel role in the pathways. In addition to its function in mitochondrial FAO, ACAD9 has been shown to have a second function as one of 14 factors responsible for assembly of complex I of the electron transport chain (ETC). -

Mitochondrial Genetics
Mitochondrial genetics Patrick Francis Chinnery and Gavin Hudson* Institute of Genetic Medicine, International Centre for Life, Newcastle University, Central Parkway, Newcastle upon Tyne NE1 3BZ, UK Introduction: In the last 10 years the field of mitochondrial genetics has widened, shifting the focus from rare sporadic, metabolic disease to the effects of mitochondrial DNA (mtDNA) variation in a growing spectrum of human disease. The aim of this review is to guide the reader through some key concepts regarding mitochondria before introducing both classic and emerging mitochondrial disorders. Sources of data: In this article, a review of the current mitochondrial genetics literature was conducted using PubMed (http://www.ncbi.nlm.nih.gov/pubmed/). In addition, this review makes use of a growing number of publically available databases including MITOMAP, a human mitochondrial genome database (www.mitomap.org), the Human DNA polymerase Gamma Mutation Database (http://tools.niehs.nih.gov/polg/) and PhyloTree.org (www.phylotree.org), a repository of global mtDNA variation. Areas of agreement: The disruption in cellular energy, resulting from defects in mtDNA or defects in the nuclear-encoded genes responsible for mitochondrial maintenance, manifests in a growing number of human diseases. Areas of controversy: The exact mechanisms which govern the inheritance of mtDNA are hotly debated. Growing points: Although still in the early stages, the development of in vitro genetic manipulation could see an end to the inheritance of the most severe mtDNA disease. Keywords: mitochondria/genetics/mitochondrial DNA/mitochondrial disease/ mtDNA Accepted: April 16, 2013 Mitochondria *Correspondence address. The mitochondrion is a highly specialized organelle, present in almost all Institute of Genetic Medicine, International eukaryotic cells and principally charged with the production of cellular Centre for Life, Newcastle energy through oxidative phosphorylation (OXPHOS). -
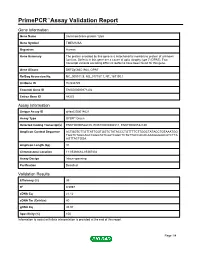
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name transmembrane protein 126A Gene Symbol TMEM126A Organism Human Gene Summary The protein encoded by this gene is a mitochondrial membrane protein of unknown function. Defects in this gene are a cause of optic atrophy type 7 (OPA7). Two transcript variants encoding different isoforms have been found for this gene. Gene Aliases DKFZp586C1924, OPA7 RefSeq Accession No. NC_000011.9, NG_017157.1, NT_167190.1 UniGene ID Hs.533725 Ensembl Gene ID ENSG00000171202 Entrez Gene ID 84233 Assay Information Unique Assay ID qHsaCID0014621 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000528105, ENST00000304511, ENST00000532180 Amplicon Context Sequence ACTGGTCTTGTTATTGGTGGTCTATACCCTGTTTTCTTGGCTATACCTGTAAATGG TGGTCTAGCAGCCAGGTATCAATCAGCTCTGTTACCACACAAAGGGAACATCTTA AGTTACTGGA Amplicon Length (bp) 91 Chromosome Location 11:85366682-85367402 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 99 R2 0.9997 cDNA Cq 21.12 cDNA Tm (Celsius) 80 gDNA Cq 35.93 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report TMEM126A, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Human Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. -

TMEM126B (BC017574) Human Untagged Clone – SC105901
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for SC105901 TMEM126B (BC017574) Human Untagged Clone Product data: Product Type: Expression Plasmids Product Name: TMEM126B (BC017574) Human Untagged Clone Tag: Tag Free Symbol: TMEM126B Synonyms: HT007; MGC111203; transmembrane protein 126B Vector: pCMV6-XL5 E. coli Selection: Ampicillin (100 ug/mL) Cell Selection: None Fully Sequenced ORF: >NCBI ORF sequence for BC017574, the custom clone sequence may differ by one or more nucleotides ATGGCAGCATCTATGCATGGTCAGCCCAGTCCTTCTCTAGAAGATGCAAAACTCAGAAGACCAATGGTCA TAGAAATCATAGAAAAAAATTTTGACTATCTTAGAAAAGAAATGACACAAAATATATATCAAATGGCGAC ATTTGGAACAACAGCTGGTTTCTCTGGAATATTCTCAAACTTCCTGTTCAGACGCTGCTTCAAGGTTAAA CATGATGCTTTGAAGACATATGCATCATTGGCTACACTTCCATTTTTGTCTACTGTTGTTACTGACAAGC TTTTTGTAATTGATGCTTTGTATTCAGGTGAATTTAAATTCACTAATGTATAA 5' Read Nucleotide >OriGene 5' read for BC017574 unedited Sequence: GTAATACGACTCACTATAGGGCGGCCGCGAATTCGGCACGAGGCCAAAAGGTGGTGTTCG GGTATGAGGCTGGGACTAAGCCAAGGGATTCAGGTGTGGTGCCGGTGGGAACTGAGGAAG CGCCCAAGGAAATGAAACACGATTTCCAAAATGAACTTAATCTTTCATGAGAAACTGAGG ATAGAGATGTCAATAAGCAGCCACTGTTTCCACCTCCCCACCTGAAGAGCTAGGAGGACA ACTACAAAGAGCCTGACTGCCTTCTCGGAATGAGGAGAGAGGAAAACAGCAACAGTATCA GTTTTCAAGATGGCAGCATCTATGCATGGTCAGCCCAGTCCTTCTCTAGAAGATGCAAAA CTCAGAAGACCAATGGTCATAGAAATCATAGAAAAAAATTTTGACTATCTTAGAAAAGAA ATGACACAAAATATATATCAAATGGCGACATTTGGAACAACAGCTGGTTTCTCTGGAATA TTCTCAAACTTCCTGTTCAGACGCTGCTTCAAGGTTAAACATGATGCTTTGAAGACATAT -

Bayesian Hidden Markov Tree Models for Clustering Genes with Shared Evolutionary History
Submitted to the Annals of Applied Statistics arXiv: arXiv:0000.0000 BAYESIAN HIDDEN MARKOV TREE MODELS FOR CLUSTERING GENES WITH SHARED EVOLUTIONARY HISTORY By Yang Liy,{,∗, Shaoyang Ningy,∗, Sarah E. Calvoz,x,{, Vamsi K. Moothak,z,x,{ and Jun S. Liuy Harvard Universityy, Broad Institutez, Harvard Medical Schoolx, Massachusetts General Hospital{, and Howard Hughes Medical Institutek Determination of functions for poorly characterized genes is cru- cial for understanding biological processes and studying human dis- eases. Functionally associated genes are often gained and lost together through evolution. Therefore identifying co-evolution of genes can predict functional gene-gene associations. We describe here the full statistical model and computational strategies underlying the orig- inal algorithm CLustering by Inferred Models of Evolution (CLIME 1.0) recently reported by us [Li et al., 2014]. CLIME 1.0 employs a mixture of tree-structured hidden Markov models for gene evolution process, and a Bayesian model-based clustering algorithm to detect gene modules with shared evolutionary histories (termed evolutionary conserved modules, or ECMs). A Dirichlet process prior was adopted for estimating the number of gene clusters and a Gibbs sampler was developed for posterior sampling. We further developed an extended version, CLIME 1.1, to incorporate the uncertainty on the evolution- ary tree structure. By simulation studies and benchmarks on real data sets, we show that CLIME 1.0 and CLIME 1.1 outperform traditional methods that use simple metrics (e.g., the Hamming distance or Pear- son correlation) to measure co-evolution between pairs of genes. 1. Introduction. The human genome encodes more than 20,000 protein- coding genes, of which a large fraction do not have annotated function to date [Galperin and Koonin, 2010].