CNKSR1 Gene Defect Can Cause Syndromic Autosomal Recessive Intellectual Disability
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Noelia Díaz Blanco
Effects of environmental factors on the gonadal transcriptome of European sea bass (Dicentrarchus labrax), juvenile growth and sex ratios Noelia Díaz Blanco Ph.D. thesis 2014 Submitted in partial fulfillment of the requirements for the Ph.D. degree from the Universitat Pompeu Fabra (UPF). This work has been carried out at the Group of Biology of Reproduction (GBR), at the Department of Renewable Marine Resources of the Institute of Marine Sciences (ICM-CSIC). Thesis supervisor: Dr. Francesc Piferrer Professor d’Investigació Institut de Ciències del Mar (ICM-CSIC) i ii A mis padres A Xavi iii iv Acknowledgements This thesis has been made possible by the support of many people who in one way or another, many times unknowingly, gave me the strength to overcome this "long and winding road". First of all, I would like to thank my supervisor, Dr. Francesc Piferrer, for his patience, guidance and wise advice throughout all this Ph.D. experience. But above all, for the trust he placed on me almost seven years ago when he offered me the opportunity to be part of his team. Thanks also for teaching me how to question always everything, for sharing with me your enthusiasm for science and for giving me the opportunity of learning from you by participating in many projects, collaborations and scientific meetings. I am also thankful to my colleagues (former and present Group of Biology of Reproduction members) for your support and encouragement throughout this journey. To the “exGBRs”, thanks for helping me with my first steps into this world. Working as an undergrad with you Dr. -
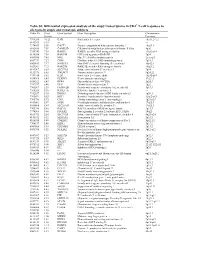
Table S1| Differential Expression Analysis of the Atopy Transcriptome
Table S1| Differential expression analysis of the atopy transcriptome in CD4+ T-cell responses to allergens in atopic and nonatopic subjects Probe ID S.test Gene Symbol Gene Description Chromosome Statistic Location 7994280 10.32 IL4R Interleukin 4 receptor 16p11.2-12.1 8143383 8.95 --- --- --- 7974689 8.50 DACT1 Dapper, antagonist of beta-catenin, homolog 1 14q23.1 8102415 7.59 CAMK2D Calcium/calmodulin-dependent protein kinase II delta 4q26 7950743 7.58 RAB30 RAB30, member RAS oncogene family 11q12-q14 8136580 7.54 RAB19B GTP-binding protein RAB19B 7q34 8043504 7.45 MAL Mal, T-cell differentiation protein 2cen-q13 8087739 7.27 CISH Cytokine inducible SH2-containing protein 3p21.3 8000413 7.17 NSMCE1 Non-SMC element 1 homolog (S. cerevisiae) 16p12.1 8021301 7.15 RAB27B RAB27B, member RAS oncogene family 18q21.2 8143367 6.83 SLC37A3 Solute carrier family 37 member 3 7q34 8152976 6.65 TMEM71 Transmembrane protein 71 8q24.22 7931914 6.56 IL2R Interleukin 2 receptor, alpha 10p15-p14 8014768 6.43 PLXDC1 Plexin domain containing 1 17q21.1 8056222 6.43 DPP4 Dipeptidyl-peptidase 4 (CD26) 2q24.3 7917697 6.40 GFI1 Growth factor independent 1 1p22 7903507 6.39 FAM102B Family with sequence similarity 102, member B 1p13.3 7968236 5.96 RASL11A RAS-like, family 11, member A --- 7912537 5.95 DHRS3 Dehydrogenase/reductase (SDR family) member 3 1p36.1 7963491 5.83 KRT1 Keratin 1 (epidermolytic hyperkeratosis) 12q12-q13 7903786 5.72 CSF1 Colony stimulating factor 1 (macrophage) 1p21-p13 8019061 5.67 SGSH N-sulfoglucosamine sulfohydrolase (sulfamidase) 17q25.3 -

X-Linked Diseases: Susceptible Females
REVIEW ARTICLE X-linked diseases: susceptible females Barbara R. Migeon, MD 1 The role of X-inactivation is often ignored as a prime cause of sex data include reasons why women are often protected from the differences in disease. Yet, the way males and females express their deleterious variants carried on their X chromosome, and the factors X-linked genes has a major role in the dissimilar phenotypes that that render women susceptible in some instances. underlie many rare and common disorders, such as intellectual deficiency, epilepsy, congenital abnormalities, and diseases of the Genetics in Medicine (2020) 22:1156–1174; https://doi.org/10.1038/s41436- heart, blood, skin, muscle, and bones. Summarized here are many 020-0779-4 examples of the different presentations in males and females. Other INTRODUCTION SEX DIFFERENCES ARE DUE TO X-INACTIVATION Sex differences in human disease are usually attributed to The sex differences in the effect of X-linked pathologic variants sex specific life experiences, and sex hormones that is due to our method of X chromosome dosage compensation, influence the function of susceptible genes throughout the called X-inactivation;9 humans and most placental mammals – genome.1 5 Such factors do account for some dissimilarities. compensate for the sex difference in number of X chromosomes However, a major cause of sex-determined expression of (that is, XX females versus XY males) by transcribing only one disease has to do with differences in how males and females of the two female X chromosomes. X-inactivation silences all X transcribe their gene-rich human X chromosomes, which is chromosomes but one; therefore, both males and females have a often underappreciated as a cause of sex differences in single active X.10,11 disease.6 Males are the usual ones affected by X-linked For 46 XY males, that X is the only one they have; it always pathogenic variants.6 Females are biologically superior; a comes from their mother, as fathers contribute their Y female usually has no disease, or much less severe disease chromosome. -

The Landscape of Human Mutually Exclusive Splicing
bioRxiv preprint doi: https://doi.org/10.1101/133215; this version posted May 2, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. The landscape of human mutually exclusive splicing Klas Hatje1,2,#,*, Ramon O. Vidal2,*, Raza-Ur Rahman2, Dominic Simm1,3, Björn Hammesfahr1,$, Orr Shomroni2, Stefan Bonn2§ & Martin Kollmar1§ 1 Group of Systems Biology of Motor Proteins, Department of NMR-based Structural Biology, Max-Planck-Institute for Biophysical Chemistry, Göttingen, Germany 2 Group of Computational Systems Biology, German Center for Neurodegenerative Diseases, Göttingen, Germany 3 Theoretical Computer Science and Algorithmic Methods, Institute of Computer Science, Georg-August-University Göttingen, Germany § Corresponding authors # Current address: Roche Pharmaceutical Research and Early Development, Pharmaceutical Sciences, Roche Innovation Center Basel, F. Hoffmann-La Roche Ltd., Basel, Switzerland $ Current address: Research and Development - Data Management (RD-DM), KWS SAAT SE, Einbeck, Germany * These authors contributed equally E-mail addresses: KH: [email protected], RV: [email protected], RR: [email protected], DS: [email protected], BH: [email protected], OS: [email protected], SB: [email protected], MK: [email protected] - 1 - bioRxiv preprint doi: https://doi.org/10.1101/133215; this version posted May 2, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Supplementary Table 2
Supplementary Table 2. Differentially Expressed Genes following Sham treatment relative to Untreated Controls Fold Change Accession Name Symbol 3 h 12 h NM_013121 CD28 antigen Cd28 12.82 BG665360 FMS-like tyrosine kinase 1 Flt1 9.63 NM_012701 Adrenergic receptor, beta 1 Adrb1 8.24 0.46 U20796 Nuclear receptor subfamily 1, group D, member 2 Nr1d2 7.22 NM_017116 Calpain 2 Capn2 6.41 BE097282 Guanine nucleotide binding protein, alpha 12 Gna12 6.21 NM_053328 Basic helix-loop-helix domain containing, class B2 Bhlhb2 5.79 NM_053831 Guanylate cyclase 2f Gucy2f 5.71 AW251703 Tumor necrosis factor receptor superfamily, member 12a Tnfrsf12a 5.57 NM_021691 Twist homolog 2 (Drosophila) Twist2 5.42 NM_133550 Fc receptor, IgE, low affinity II, alpha polypeptide Fcer2a 4.93 NM_031120 Signal sequence receptor, gamma Ssr3 4.84 NM_053544 Secreted frizzled-related protein 4 Sfrp4 4.73 NM_053910 Pleckstrin homology, Sec7 and coiled/coil domains 1 Pscd1 4.69 BE113233 Suppressor of cytokine signaling 2 Socs2 4.68 NM_053949 Potassium voltage-gated channel, subfamily H (eag- Kcnh2 4.60 related), member 2 NM_017305 Glutamate cysteine ligase, modifier subunit Gclm 4.59 NM_017309 Protein phospatase 3, regulatory subunit B, alpha Ppp3r1 4.54 isoform,type 1 NM_012765 5-hydroxytryptamine (serotonin) receptor 2C Htr2c 4.46 NM_017218 V-erb-b2 erythroblastic leukemia viral oncogene homolog Erbb3 4.42 3 (avian) AW918369 Zinc finger protein 191 Zfp191 4.38 NM_031034 Guanine nucleotide binding protein, alpha 12 Gna12 4.38 NM_017020 Interleukin 6 receptor Il6r 4.37 AJ002942 -

Exons Deletion of CNKSR2 Gene Identified in X-Linked Syndromic
Daoqi et al. BMC Medical Genetics (2020) 21:69 https://doi.org/10.1186/s12881-020-01004-2 CASE REPORT Open Access Exons deletion of CNKSR2 gene identified in X-linked syndromic intellectual disability Mei Daoqi1, Chen Guohong1, Wang Yuan1, Yang Zhixiao1, Xu Kaili1 and Mei Shiyue2* Abstract Background: The Houge type of X-linked syndromic mental retardation is an X-linked intellectual disability (XLID) recently recorded in the Online Mendelian Inheritance in Man (OMIM) and only 8 cases have been reported in literature thus far. Case presentation: We present two brothers with intractable seizures and syndromic intellectual disability with symptoms consisting of delayed development, intellectual disability, and speech and language delay. The mother was a symptomatic carrier with milder clinical phenotype. Whole exome sequencing identified a small fragment deletion spanning four exons, about 9.5 kilobases (kb) in length in the CNKSR2 gene in the patients. The mutation co-segregation revealed that exon deletions occurred de novo in the proband’s mother. Conclusion: Although large deletions have been reported, no small deletions have yet been identified. In this case report, we identified a small deletion in the CNKSR2 gene. This study enhances our knowledge of the CNKSR2 gene mutation spectrum and provides further information about the phenotypic characteristics of X-linked syndromic intellectual disability. Keywords: X-linked syndromic mental retardation, Intractable seizures, CNKSR2 gene, Exons deletion Background (#301008) in 2017. MRXSHG is caused by a deficiency X-linked intellectual disability (XLID), also known as of the CNKSR2 gene and only 8 cases have been X-linked mental retardation (XLMR), refers to a gener- reported in literature. -

Molecular Signatures Differentiate Immune States in Type 1 Diabetes Families
Page 1 of 65 Diabetes Molecular signatures differentiate immune states in Type 1 diabetes families Yi-Guang Chen1, Susanne M. Cabrera1, Shuang Jia1, Mary L. Kaldunski1, Joanna Kramer1, Sami Cheong2, Rhonda Geoffrey1, Mark F. Roethle1, Jeffrey E. Woodliff3, Carla J. Greenbaum4, Xujing Wang5, and Martin J. Hessner1 1The Max McGee National Research Center for Juvenile Diabetes, Children's Research Institute of Children's Hospital of Wisconsin, and Department of Pediatrics at the Medical College of Wisconsin Milwaukee, WI 53226, USA. 2The Department of Mathematical Sciences, University of Wisconsin-Milwaukee, Milwaukee, WI 53211, USA. 3Flow Cytometry & Cell Separation Facility, Bindley Bioscience Center, Purdue University, West Lafayette, IN 47907, USA. 4Diabetes Research Program, Benaroya Research Institute, Seattle, WA, 98101, USA. 5Systems Biology Center, the National Heart, Lung, and Blood Institute, the National Institutes of Health, Bethesda, MD 20824, USA. Corresponding author: Martin J. Hessner, Ph.D., The Department of Pediatrics, The Medical College of Wisconsin, Milwaukee, WI 53226, USA Tel: 011-1-414-955-4496; Fax: 011-1-414-955-6663; E-mail: [email protected]. Running title: Innate Inflammation in T1D Families Word count: 3999 Number of Tables: 1 Number of Figures: 7 1 For Peer Review Only Diabetes Publish Ahead of Print, published online April 23, 2014 Diabetes Page 2 of 65 ABSTRACT Mechanisms associated with Type 1 diabetes (T1D) development remain incompletely defined. Employing a sensitive array-based bioassay where patient plasma is used to induce transcriptional responses in healthy leukocytes, we previously reported disease-specific, partially IL-1 dependent, signatures associated with pre and recent onset (RO) T1D relative to unrelated healthy controls (uHC). -

Stargazin and Other Transmembrane AMPA Receptor Regulating Proteins Interact with Synaptic Scaffolding Protein MAGI-2 in Brain
The Journal of Neuroscience, July 26, 2006 • 26(30):7875–7884 • 7875 Cellular/Molecular Stargazin and Other Transmembrane AMPA Receptor Regulating Proteins Interact with Synaptic Scaffolding Protein MAGI-2 in Brain Fang Deng, Maureen G. Price, Caleb F. Davis, Mayra Mori, and Daniel L. Burgess Department of Neurology, Baylor College of Medicine, Houston, Texas 77030 The spatial coordination of neurotransmitter receptors with other postsynaptic signaling and structural molecules is regulated by a diverse array of cell-specific scaffolding proteins. The synaptic trafficking of AMPA receptors by the stargazin protein in some neurons, for example, depends on specific interactions between the C terminus of stargazin and the PDZ [postsynaptic density-95 (PSD-95)/Discs large/zona occludens-1] domains of membrane-associated guanylate kinase scaffolding proteins PSD-93 or PSD-95. Stargazin [Cacng2 (Ca 2ϩ channel ␥2 subunit)] is one of four closely related proteins recently categorized as transmembrane AMPA receptor regulating proteins (TARPs) that appear to share similar functions but exhibit distinct expression patterns in the CNS. We used yeast two-hybrid screeningtoidentifyMAGI-2(membraneassociatedguanylatekinase,WWandPDZdomaincontaining2)asanovelcandidateinteractor with the cytoplasmic C termini of the TARPs. MAGI-2 [also known as S-SCAM (synaptic scaffolding molecule)] is a multi-PDZ domain scaffolding protein that interacts with several different ligands in brain, including PTEN (phosphatase and tensin homolog), dasm1 (dendrite arborization and synapse maturation 1), dendrin, axin, - and ␦-catenin, neuroligin, hyperpolarization-activated cation channels, 1-adrenergic receptors, and NMDA receptors. We confirmed that MAGI-2 coimmunoprecipitated with stargazin in vivo from mouse cerebral cortex and used in vitro assays to localize the interaction to the C-terminal -TTPV amino acid motif of stargazin and the PDZ1, PDZ3, and PDZ5 domains of MAGI-2. -

Development of Novel Analysis and Data Integration Systems to Understand Human Gene Regulation
Development of novel analysis and data integration systems to understand human gene regulation Dissertation zur Erlangung des Doktorgrades Dr. rer. nat. der Fakult¨atf¨urMathematik und Informatik der Georg-August-Universit¨atG¨ottingen im PhD Programme in Computer Science (PCS) der Georg-August University School of Science (GAUSS) vorgelegt von Raza-Ur Rahman aus Pakistan G¨ottingen,April 2018 Prof. Dr. Stefan Bonn, Zentrum f¨urMolekulare Neurobiologie (ZMNH), Betreuungsausschuss: Institut f¨urMedizinische Systembiologie, Hamburg Prof. Dr. Tim Beißbarth, Institut f¨urMedizinische Statistik, Universit¨atsmedizin, Georg-August Universit¨at,G¨ottingen Prof. Dr. Burkhard Morgenstern, Institut f¨urMikrobiologie und Genetik Abtl. Bioinformatik, Georg-August Universit¨at,G¨ottingen Pr¨ufungskommission: Prof. Dr. Stefan Bonn, Zentrum f¨urMolekulare Neurobiologie (ZMNH), Referent: Institut f¨urMedizinische Systembiologie, Hamburg Prof. Dr. Tim Beißbarth, Institut f¨urMedizinische Statistik, Universit¨atsmedizin, Korreferent: Georg-August Universit¨at,G¨ottingen Prof. Dr. Burkhard Morgenstern, Weitere Mitglieder Institut f¨urMikrobiologie und Genetik Abtl. Bioinformatik, der Pr¨ufungskommission: Georg-August Universit¨at,G¨ottingen Prof. Dr. Carsten Damm, Institut f¨urInformatik, Georg-August Universit¨at,G¨ottingen Prof. Dr. Florentin W¨org¨otter, Physikalisches Institut Biophysik, Georg-August-Universit¨at,G¨ottingen Prof. Dr. Stephan Waack, Institut f¨urInformatik, Georg-August Universit¨at,G¨ottingen Tag der m¨undlichen Pr¨ufung: der 30. M¨arz2018 -

A Network Medicine Approach for Drug Repurposing in Duchenne Muscular Dystrophy
G C A T T A C G G C A T genes Article A Network Medicine Approach for Drug Repurposing in Duchenne Muscular Dystrophy Salvo Danilo Lombardo 1 , Maria Sofia Basile 2 , Rosella Ciurleo 2, Alessia Bramanti 2, Antonio Arcidiacono 3, Katia Mangano 3 , Placido Bramanti 2, Ferdinando Nicoletti 3,* and Paolo Fagone 3 1 Department of Structural & Computational Biology at the Max Perutz Labs, University of Vienna, 1010 Vienna, Austria; [email protected] 2 IRCCS Centro Neurolesi “Bonino-Pulejo”, Via Provinciale Palermo, Contrada Casazza, 98124 Messina, Italy; sofi[email protected] (M.S.B.); [email protected] (R.C.); [email protected] (A.B.); [email protected] (P.B.) 3 Department of Biomedical and Biotechnological Sciences, University of Catania, Via S. Sofia 89, 95123 Catania, Italy; [email protected] (A.A.); [email protected] (K.M.); [email protected] (P.F.) * Correspondence: [email protected] Abstract: Duchenne muscular dystrophy (DMD) is a progressive hereditary muscular disease caused by a lack of dystrophin, leading to membrane instability, cell damage, and inflammatory response. However, gene-editing alone is not enough to restore the healthy phenotype and additional treat- ments are required. In the present study, we have first conducted a meta-analysis of three microarray datasets, GSE38417, GSE3307, and GSE6011, to identify the differentially expressed genes (DEGs) be- tween healthy donors and DMD patients. We have then integrated this analysis with the knowledge Citation: Lombardo, S.D.; Basile, obtained from DisGeNET and DIAMOnD, a well-known algorithm for drug–gene association discov- M.S.; Ciurleo, R.; Bramanti, A.; eries in the human interactome. -

CNKSR2 Deletions: a Novel Cause of X-Linked Intellectual Disability and Seizures Umut Aypar,1 Elaine C
RESEARCH LETTER CNKSR2 Deletions: A Novel Cause of X-Linked Intellectual Disability and Seizures Umut Aypar,1 Elaine C. Wirrell,2 and Nicole L. Hoppman1* 1Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, Minnesota 2Department of Neurology, Mayo Clinic, Rochester, Minnesota Manuscript Received: 9 May 2014; Manuscript Accepted: 12 November 2014 TO THE EDITOR X-linked intellectual disability (XLID) accounts for approximately How to Cite this Article: 10% of intellectual disability in males and contributes to the excess Aypar U, Wirrell E.C, Hoppman N.L. 2015. of males in the intellectually disabled population (male to female CNKSR2 Deletions: A Novel Cause of X- ratio 1.3–1.4 to 1) [Ropers and Hamel, 2005]. Underlying causes of Linked Intellectual Disability and Seizures. XLID have been extensively studied in recent years, and as a result Am J Med Genet Part A. 9999:1–3. mutations causing XLID have been described in about 106 genes [Piton et al., 2013]. Recently, Houge et al. [2012] described a maternally inherited 234 kilobase deletion on the X chromosome, which removed the majority of the CNKSR2 gene (OMIM #300724; He sat alone at approximately one year and walked alone at 21,375,312 – 21,609,484, genome build hg19; Fig. 1E) in a male with approximately two years. Initially, he was diagnosed with seizures developmental delay, epilepsy, and microcephaly. Since CNKSR2 at age two, which presented initially with typical events such as gene is highly expressed only in the brain [Nagase et al., 1998], staring, smacking his lips, and having episodes of whole-body Houge et al. -

University of Groningen on the Elucidation of a Tumour Suppressor
University of Groningen On the elucidation of a tumour suppressor role of 3p in lung cancer Elst, Arja ter IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite from it. Please check the document version below. Document Version Publisher's PDF, also known as Version of record Publication date: 2006 Link to publication in University of Groningen/UMCG research database Citation for published version (APA): Elst, A. T. (2006). On the elucidation of a tumour suppressor role of 3p in lung cancer. s.n. Copyright Other than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons). Take-down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons the number of authors shown on this cover page is limited to 10 maximum. Download date: 27-09-2021 Chapter 1 Candidate lung tumour suppressor regions at the short arm of chromosome 3. What evidence is there? Arja ter Elst Charles H.C.M. Buys Department of Medical Genetics, University Medical Center Groningen, Groningen, The Netherlands LUNG CANCER AND THE SHORT ARM OF CHROMOSOME 3 Lung cancer is the leading cause of cancer death among both men and women in the western world.