CNKSR2 Deletions: a Novel Cause of X-Linked Intellectual Disability and Seizures Umut Aypar,1 Elaine C
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
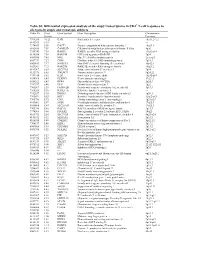
Table S1| Differential Expression Analysis of the Atopy Transcriptome
Table S1| Differential expression analysis of the atopy transcriptome in CD4+ T-cell responses to allergens in atopic and nonatopic subjects Probe ID S.test Gene Symbol Gene Description Chromosome Statistic Location 7994280 10.32 IL4R Interleukin 4 receptor 16p11.2-12.1 8143383 8.95 --- --- --- 7974689 8.50 DACT1 Dapper, antagonist of beta-catenin, homolog 1 14q23.1 8102415 7.59 CAMK2D Calcium/calmodulin-dependent protein kinase II delta 4q26 7950743 7.58 RAB30 RAB30, member RAS oncogene family 11q12-q14 8136580 7.54 RAB19B GTP-binding protein RAB19B 7q34 8043504 7.45 MAL Mal, T-cell differentiation protein 2cen-q13 8087739 7.27 CISH Cytokine inducible SH2-containing protein 3p21.3 8000413 7.17 NSMCE1 Non-SMC element 1 homolog (S. cerevisiae) 16p12.1 8021301 7.15 RAB27B RAB27B, member RAS oncogene family 18q21.2 8143367 6.83 SLC37A3 Solute carrier family 37 member 3 7q34 8152976 6.65 TMEM71 Transmembrane protein 71 8q24.22 7931914 6.56 IL2R Interleukin 2 receptor, alpha 10p15-p14 8014768 6.43 PLXDC1 Plexin domain containing 1 17q21.1 8056222 6.43 DPP4 Dipeptidyl-peptidase 4 (CD26) 2q24.3 7917697 6.40 GFI1 Growth factor independent 1 1p22 7903507 6.39 FAM102B Family with sequence similarity 102, member B 1p13.3 7968236 5.96 RASL11A RAS-like, family 11, member A --- 7912537 5.95 DHRS3 Dehydrogenase/reductase (SDR family) member 3 1p36.1 7963491 5.83 KRT1 Keratin 1 (epidermolytic hyperkeratosis) 12q12-q13 7903786 5.72 CSF1 Colony stimulating factor 1 (macrophage) 1p21-p13 8019061 5.67 SGSH N-sulfoglucosamine sulfohydrolase (sulfamidase) 17q25.3 -

X-Linked Diseases: Susceptible Females
REVIEW ARTICLE X-linked diseases: susceptible females Barbara R. Migeon, MD 1 The role of X-inactivation is often ignored as a prime cause of sex data include reasons why women are often protected from the differences in disease. Yet, the way males and females express their deleterious variants carried on their X chromosome, and the factors X-linked genes has a major role in the dissimilar phenotypes that that render women susceptible in some instances. underlie many rare and common disorders, such as intellectual deficiency, epilepsy, congenital abnormalities, and diseases of the Genetics in Medicine (2020) 22:1156–1174; https://doi.org/10.1038/s41436- heart, blood, skin, muscle, and bones. Summarized here are many 020-0779-4 examples of the different presentations in males and females. Other INTRODUCTION SEX DIFFERENCES ARE DUE TO X-INACTIVATION Sex differences in human disease are usually attributed to The sex differences in the effect of X-linked pathologic variants sex specific life experiences, and sex hormones that is due to our method of X chromosome dosage compensation, influence the function of susceptible genes throughout the called X-inactivation;9 humans and most placental mammals – genome.1 5 Such factors do account for some dissimilarities. compensate for the sex difference in number of X chromosomes However, a major cause of sex-determined expression of (that is, XX females versus XY males) by transcribing only one disease has to do with differences in how males and females of the two female X chromosomes. X-inactivation silences all X transcribe their gene-rich human X chromosomes, which is chromosomes but one; therefore, both males and females have a often underappreciated as a cause of sex differences in single active X.10,11 disease.6 Males are the usual ones affected by X-linked For 46 XY males, that X is the only one they have; it always pathogenic variants.6 Females are biologically superior; a comes from their mother, as fathers contribute their Y female usually has no disease, or much less severe disease chromosome. -

Supplementary Table 2
Supplementary Table 2. Differentially Expressed Genes following Sham treatment relative to Untreated Controls Fold Change Accession Name Symbol 3 h 12 h NM_013121 CD28 antigen Cd28 12.82 BG665360 FMS-like tyrosine kinase 1 Flt1 9.63 NM_012701 Adrenergic receptor, beta 1 Adrb1 8.24 0.46 U20796 Nuclear receptor subfamily 1, group D, member 2 Nr1d2 7.22 NM_017116 Calpain 2 Capn2 6.41 BE097282 Guanine nucleotide binding protein, alpha 12 Gna12 6.21 NM_053328 Basic helix-loop-helix domain containing, class B2 Bhlhb2 5.79 NM_053831 Guanylate cyclase 2f Gucy2f 5.71 AW251703 Tumor necrosis factor receptor superfamily, member 12a Tnfrsf12a 5.57 NM_021691 Twist homolog 2 (Drosophila) Twist2 5.42 NM_133550 Fc receptor, IgE, low affinity II, alpha polypeptide Fcer2a 4.93 NM_031120 Signal sequence receptor, gamma Ssr3 4.84 NM_053544 Secreted frizzled-related protein 4 Sfrp4 4.73 NM_053910 Pleckstrin homology, Sec7 and coiled/coil domains 1 Pscd1 4.69 BE113233 Suppressor of cytokine signaling 2 Socs2 4.68 NM_053949 Potassium voltage-gated channel, subfamily H (eag- Kcnh2 4.60 related), member 2 NM_017305 Glutamate cysteine ligase, modifier subunit Gclm 4.59 NM_017309 Protein phospatase 3, regulatory subunit B, alpha Ppp3r1 4.54 isoform,type 1 NM_012765 5-hydroxytryptamine (serotonin) receptor 2C Htr2c 4.46 NM_017218 V-erb-b2 erythroblastic leukemia viral oncogene homolog Erbb3 4.42 3 (avian) AW918369 Zinc finger protein 191 Zfp191 4.38 NM_031034 Guanine nucleotide binding protein, alpha 12 Gna12 4.38 NM_017020 Interleukin 6 receptor Il6r 4.37 AJ002942 -

Exons Deletion of CNKSR2 Gene Identified in X-Linked Syndromic
Daoqi et al. BMC Medical Genetics (2020) 21:69 https://doi.org/10.1186/s12881-020-01004-2 CASE REPORT Open Access Exons deletion of CNKSR2 gene identified in X-linked syndromic intellectual disability Mei Daoqi1, Chen Guohong1, Wang Yuan1, Yang Zhixiao1, Xu Kaili1 and Mei Shiyue2* Abstract Background: The Houge type of X-linked syndromic mental retardation is an X-linked intellectual disability (XLID) recently recorded in the Online Mendelian Inheritance in Man (OMIM) and only 8 cases have been reported in literature thus far. Case presentation: We present two brothers with intractable seizures and syndromic intellectual disability with symptoms consisting of delayed development, intellectual disability, and speech and language delay. The mother was a symptomatic carrier with milder clinical phenotype. Whole exome sequencing identified a small fragment deletion spanning four exons, about 9.5 kilobases (kb) in length in the CNKSR2 gene in the patients. The mutation co-segregation revealed that exon deletions occurred de novo in the proband’s mother. Conclusion: Although large deletions have been reported, no small deletions have yet been identified. In this case report, we identified a small deletion in the CNKSR2 gene. This study enhances our knowledge of the CNKSR2 gene mutation spectrum and provides further information about the phenotypic characteristics of X-linked syndromic intellectual disability. Keywords: X-linked syndromic mental retardation, Intractable seizures, CNKSR2 gene, Exons deletion Background (#301008) in 2017. MRXSHG is caused by a deficiency X-linked intellectual disability (XLID), also known as of the CNKSR2 gene and only 8 cases have been X-linked mental retardation (XLMR), refers to a gener- reported in literature. -

Molecular Signatures Differentiate Immune States in Type 1 Diabetes Families
Page 1 of 65 Diabetes Molecular signatures differentiate immune states in Type 1 diabetes families Yi-Guang Chen1, Susanne M. Cabrera1, Shuang Jia1, Mary L. Kaldunski1, Joanna Kramer1, Sami Cheong2, Rhonda Geoffrey1, Mark F. Roethle1, Jeffrey E. Woodliff3, Carla J. Greenbaum4, Xujing Wang5, and Martin J. Hessner1 1The Max McGee National Research Center for Juvenile Diabetes, Children's Research Institute of Children's Hospital of Wisconsin, and Department of Pediatrics at the Medical College of Wisconsin Milwaukee, WI 53226, USA. 2The Department of Mathematical Sciences, University of Wisconsin-Milwaukee, Milwaukee, WI 53211, USA. 3Flow Cytometry & Cell Separation Facility, Bindley Bioscience Center, Purdue University, West Lafayette, IN 47907, USA. 4Diabetes Research Program, Benaroya Research Institute, Seattle, WA, 98101, USA. 5Systems Biology Center, the National Heart, Lung, and Blood Institute, the National Institutes of Health, Bethesda, MD 20824, USA. Corresponding author: Martin J. Hessner, Ph.D., The Department of Pediatrics, The Medical College of Wisconsin, Milwaukee, WI 53226, USA Tel: 011-1-414-955-4496; Fax: 011-1-414-955-6663; E-mail: [email protected]. Running title: Innate Inflammation in T1D Families Word count: 3999 Number of Tables: 1 Number of Figures: 7 1 For Peer Review Only Diabetes Publish Ahead of Print, published online April 23, 2014 Diabetes Page 2 of 65 ABSTRACT Mechanisms associated with Type 1 diabetes (T1D) development remain incompletely defined. Employing a sensitive array-based bioassay where patient plasma is used to induce transcriptional responses in healthy leukocytes, we previously reported disease-specific, partially IL-1 dependent, signatures associated with pre and recent onset (RO) T1D relative to unrelated healthy controls (uHC). -

Stargazin and Other Transmembrane AMPA Receptor Regulating Proteins Interact with Synaptic Scaffolding Protein MAGI-2 in Brain
The Journal of Neuroscience, July 26, 2006 • 26(30):7875–7884 • 7875 Cellular/Molecular Stargazin and Other Transmembrane AMPA Receptor Regulating Proteins Interact with Synaptic Scaffolding Protein MAGI-2 in Brain Fang Deng, Maureen G. Price, Caleb F. Davis, Mayra Mori, and Daniel L. Burgess Department of Neurology, Baylor College of Medicine, Houston, Texas 77030 The spatial coordination of neurotransmitter receptors with other postsynaptic signaling and structural molecules is regulated by a diverse array of cell-specific scaffolding proteins. The synaptic trafficking of AMPA receptors by the stargazin protein in some neurons, for example, depends on specific interactions between the C terminus of stargazin and the PDZ [postsynaptic density-95 (PSD-95)/Discs large/zona occludens-1] domains of membrane-associated guanylate kinase scaffolding proteins PSD-93 or PSD-95. Stargazin [Cacng2 (Ca 2ϩ channel ␥2 subunit)] is one of four closely related proteins recently categorized as transmembrane AMPA receptor regulating proteins (TARPs) that appear to share similar functions but exhibit distinct expression patterns in the CNS. We used yeast two-hybrid screeningtoidentifyMAGI-2(membraneassociatedguanylatekinase,WWandPDZdomaincontaining2)asanovelcandidateinteractor with the cytoplasmic C termini of the TARPs. MAGI-2 [also known as S-SCAM (synaptic scaffolding molecule)] is a multi-PDZ domain scaffolding protein that interacts with several different ligands in brain, including PTEN (phosphatase and tensin homolog), dasm1 (dendrite arborization and synapse maturation 1), dendrin, axin, - and ␦-catenin, neuroligin, hyperpolarization-activated cation channels, 1-adrenergic receptors, and NMDA receptors. We confirmed that MAGI-2 coimmunoprecipitated with stargazin in vivo from mouse cerebral cortex and used in vitro assays to localize the interaction to the C-terminal -TTPV amino acid motif of stargazin and the PDZ1, PDZ3, and PDZ5 domains of MAGI-2. -

Perkinelmer Genomics to Request the Saliva Swab Collection Kit for Patients That Cannot Provide a Blood Sample As Whole Blood Is the Preferred Sample
Autism and Intellectual Disability TRIO Panel Test Code TR002 Test Summary This test analyzes 2429 genes that have been associated with Autism and Intellectual Disability and/or disorders associated with Autism and Intellectual Disability with the analysis being performed as a TRIO Turn-Around-Time (TAT)* 3 - 5 weeks Acceptable Sample Types Whole Blood (EDTA) (Preferred sample type) DNA, Isolated Dried Blood Spots Saliva Acceptable Billing Types Self (patient) Payment Institutional Billing Commercial Insurance Indications for Testing Comprehensive test for patients with intellectual disability or global developmental delays (Moeschler et al 2014 PMID: 25157020). Comprehensive test for individuals with multiple congenital anomalies (Miller et al. 2010 PMID 20466091). Patients with autism/autism spectrum disorders (ASDs). Suspected autosomal recessive condition due to close familial relations Previously negative karyotyping and/or chromosomal microarray results. Test Description This panel analyzes 2429 genes that have been associated with Autism and ID and/or disorders associated with Autism and ID. Both sequencing and deletion/duplication (CNV) analysis will be performed on the coding regions of all genes included (unless otherwise marked). All analysis is performed utilizing Next Generation Sequencing (NGS) technology. CNV analysis is designed to detect the majority of deletions and duplications of three exons or greater in size. Smaller CNV events may also be detected and reported, but additional follow-up testing is recommended if a smaller CNV is suspected. All variants are classified according to ACMG guidelines. Condition Description Autism Spectrum Disorder (ASD) refers to a group of developmental disabilities that are typically associated with challenges of varying severity in the areas of social interaction, communication, and repetitive/restricted behaviors. -

The Discovery of Integrated Gene Networks for Autism and Related Disorders
Downloaded from genome.cshlp.org on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press The discovery of integrated gene networks for autism and related disorders Fereydoun Hormozdiari1*, Osnat Penn1*, Elhanan Borenstein1, Evan. E. Eichler1, 2 * Joint First Authors 1 Department of Genome Sciences, University of Washington, Seattle, WA 98195 2 Howard Hughes Medical Institute, University of Washington, Seattle, WA 98195 Correspondence to: Evan Eichler, PhD Department of Genome Sciences University of Washington School of Medicine Foege S-413A, Box 355065 3720 15th Ave NE Seattle, WA 98195 Email: [email protected] 1 Downloaded from genome.cshlp.org on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press ABSTRACT Despite considerable genetic heterogeneity underlying neurodevelopmental diseases, there is compelling evidence that many disease genes will map to a much smaller number of biological subnetworks. We developed a computational method, termed MAGI (Merging Affected Genes into Integrated-networks), that simultaneously integrates protein-protein interaction and RNA- seq expression profiles during brain development to discover “modules” enriched for de novo mutations in probands. We applied this method to recent exome sequencing of 1116 autism and intellectual disability patients discovering two distinct modules that differ in their properties and associated phenotypes. The first module consists of 80 genes associated with Wnt, Notch, SWI/SNF and NCOR complexes and shows the highest expression early during embryonic development (8–16 post-conception weeks, pcw). The second module consists of 24 genes associated with synaptic function, including long-term potentiation and calcium signaling with higher levels of postnatal expression. Patients with de novo mutations in these modules are more significantly intellectually impaired and carry more severe missense mutations when compared to probands with de novo mutations outside of these modules. -

Interaction Networks of Lithium and Valproate Molecular Targets Reveal a Striking Enrichment of Apoptosis Functional Clusters and Neurotrophin Signaling
The Pharmacogenomics Journal (2012) 12, 328–341 & 2012 Macmillan Publishers Limited. All rights reserved 1470-269X/12 www.nature.com/tpj ORIGINAL ARTICLE Interaction networks of lithium and valproate molecular targets reveal a striking enrichment of apoptosis functional clusters and neurotrophin signaling A Gupta1, TG Schulze2, The overall neurobiological mechanisms by which lithium and valproate 3 1 stabilize mood in bipolar disorder patients have yet to be fully defined. The V Nagarajan , N Akula , therapeutic efficacy and dissimilar chemical structures of these medications 1 1 W Corona , X-y Jiang , suggest that they perturb both shared and disparate cellular processes. To N Hunter1, FJ McMahon1 and investigate key pathways and functional clusters involved in the global action SD Detera-Wadleigh1 of lithium and valproate, we generated interaction networks formed by well- supported drug targets. Striking functional similarities emerged. Intersecting 1Human Genetics Branch, National Institute of nodes in lithium and valproate networks highlighted a strong enrichment Mental Health, National Institutes of Health, of apoptosis clusters and neurotrophin signaling. Other enriched pathways 2 Bethesda, MD, USA; Department of Psychiatry included MAPK, ErbB, insulin, VEGF, Wnt and long-term potentiation and Psychotherapy, University Medical Center, Georg-August-Universita¨t, Go¨ttingen, Germany indicating a widespread effect of both drugs on diverse signaling systems. and 3Bioinformatics and Computational MAPK1/3 and AKT1/2 were the most preponderant nodes across pathways Biosciences Branch, Office of Cyber Infrastructure suggesting a central role in mediating pathway interactions. The conver- and Computational Biology, National Institute of gence of biological responses unveils a functional signature for lithium and Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, USA valproate that could be key modulators of their therapeutic efficacy. -

Supplementary Figures
Supplementary Figures Fig. S1. Distribution of total mutation pool. a) Statistics of different mutational types. b) The distribution of mutational allele frequency. c) Circle graph illustrating the chromosomal assignment of total mutation pool. The outer ring provides an overview of detected ctDNA mutations in each chromosome, while the inner ring shows the mutation burden of each chromosome. 1 Fig. S2. DNMT3A mutations in the experimental cohort. The upper graph shows all 19 DNMT3A mutations and their protein localization. The red rectangle indicates DNA_methylase domains. The lollipop graph is generated by online tools of cbioportal (http://www.cbioportal.org/visualize). The lower graph illustrates the diagnostic age distribution for patients with and without DNMT3A mutations. The p value is calculated by nonparametric t-test. 2 Fig. S3. Overview of clinical actionability for patients with drug-sensitive and resistant mutations. Each horizontal column represents an individual patients. Columns with different colors indicate diverse actionable mutations, and the length of column represents the number of mutations. Patients are divided into three groups using horizontal dotted lines, according to the mutation types. 3 Fig. S4. Concordance and mutant allele frequencies for matched sample pairs. Venn diagrams present the mutation overlap between matched tissue and blood samples. Scatter diagrams show the correlation between the allele frequencies of overlapping mutations in ctDNA vs. tissue DNA. Each dot represents an overlapping mutation, -

CNKSR2 (NM 001168649) Human Tagged ORF Clone Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RC230540 CNKSR2 (NM_001168649) Human Tagged ORF Clone Product data: Product Type: Expression Plasmids Product Name: CNKSR2 (NM_001168649) Human Tagged ORF Clone Tag: Myc-DDK Symbol: CNKSR2 Synonyms: CNK2; KSR2; MAGUIN; MRXSHG Vector: pCMV6-Entry (PS100001) E. coli Selection: Kanamycin (25 ug/mL) Cell Selection: Neomycin This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 5 CNKSR2 (NM_001168649) Human Tagged ORF Clone – RC230540 ORF Nucleotide >RC230540 representing NM_001168649 Sequence: Red=Cloning site Blue=ORF Green=Tags(s) TTTTGTAATACGACTCACTATAGGGCGGCCGGGAATTCGTCGACTGGATCCGGTACCGAGGAGATCTGCC GCCGCGATCGCC ATGGCTCTGATAATGGAACCGGTGAGCAAATGGTCTCCGAGTCAAGTAGTGGACTGGATGAAAGGTCTTG ATGACTGTTTGCAGCAGTATATTAAGAACTTTGAGAGGGAGAAGATCAGTGGGGACCAGCTGCTGCGCAT TACACATCAGGAGCTAGAAGATCTGGGGGTCAGCCGCATTGGCCATCAGGAACTGATCTTGGAAGCAGTT GACCTTCTGTGTGCATTGAATTATGGCTTGGAAACAGAAAATCTAAAAACCCTTTCTCACAAGTTGAATG CATCTGCCAAAAATCTGCAGAATTTTATAACAGGAAGGAGAAGGAGTGGCCATTATGATGGGAGGACCAG CCGAAAATTGCCAAACGACTTTCTGACCTCAGTTGTGGATCTGATTGGAGCAGCCAAGAGTCTGCTTGCC TGGTTGGACAGGTCACCATTTGCTGCTGTGACAGACTATTCAGTTACAAGAAATAATGTCATACAACTCT GCCTGGAGTTAACAACAATTGTGCAACAGGATTGTACTGTATATGAAACAGAGAATAAAATTCTTCACGT GTGTAAAACTCTTTCTGGAGTCTGTGACCACATCATATCCCTGTCGTCAGATCCTCTGGTTTCACAGTCT -

Multiplex Gene and Phenotype Network to Characterize Shared Genetic Pathways of Epilepsy and Autism Jacqueline Peng1,2, Yunyun Zhou2 & Kai Wang2,3*
www.nature.com/scientificreports OPEN Multiplex gene and phenotype network to characterize shared genetic pathways of epilepsy and autism Jacqueline Peng1,2, Yunyun Zhou2 & Kai Wang2,3* It is well established that epilepsy and autism spectrum disorder (ASD) commonly co-occur; however, the underlying biological mechanisms of the co-occurence from their genetic susceptibility are not well understood. Our aim in this study is to characterize genetic modules of subgroups of epilepsy and autism genes that have similar phenotypic manifestations and biological functions. We frst integrate a large number of expert-compiled and well-established epilepsy- and ASD-associated genes in a multiplex network, where one layer is connected through protein–protein interaction (PPI) and the other layer through gene-phenotype associations. We identify two modules in the multiplex network, which are signifcantly enriched in genes associated with both epilepsy and autism as well as genes highly expressed in brain tissues. We fnd that the frst module, which represents the Gene Ontology category of ion transmembrane transport, is more epilepsy-focused, while the second module, representing synaptic signaling, is more ASD-focused. However, because of their enrichment in common genes and association with both epilepsy and ASD phenotypes, these modules point to genetic etiologies and biological processes shared between specifc subtypes of epilepsy and ASD. Finally, we use our analysis to prioritize new candidate genes for epilepsy (i.e. ANK2, CACNA1E, CACNA2D3, GRIA2, DLG4) for further validation. The analytical approaches in our study can be applied to similar studies in the future to investigate the genetic connections between diferent human diseases.