Network-Based Prediction of Polygenic Disease Genes Involved in Cell Motility Miriam Bern†, Alexander King†, Derek A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Downloads/ (Accessed on 17 January 2020)
cells Review Novel Approaches for Identifying the Molecular Background of Schizophrenia Arkadiy K. Golov 1,2,*, Nikolay V. Kondratyev 1 , George P. Kostyuk 3 and Vera E. Golimbet 1 1 Mental Health Research Center, 34 Kashirskoye shosse, 115522 Moscow, Russian; [email protected] (N.V.K.); [email protected] (V.E.G.) 2 Institute of Gene Biology, Russian Academy of Sciences, 34/5 Vavilova Street, 119334 Moscow, Russian 3 Alekseev Psychiatric Clinical Hospital No. 1, 2 Zagorodnoye shosse, 115191 Moscow, Russian; [email protected] * Correspondence: [email protected] Received: 5 November 2019; Accepted: 16 January 2020; Published: 18 January 2020 Abstract: Recent advances in psychiatric genetics have led to the discovery of dozens of genomic loci associated with schizophrenia. However, a gap exists between the detection of genetic associations and understanding the underlying molecular mechanisms. This review describes the basic approaches used in the so-called post-GWAS studies to generate biological interpretation of the existing population genetic data, including both molecular (creation and analysis of knockout animals, exploration of the transcriptional effects of common variants in human brain cells) and computational (fine-mapping of causal variability, gene set enrichment analysis, partitioned heritability analysis) methods. The results of the crucial studies, in which these approaches were used to uncover the molecular and neurobiological basis of the disease, are also reported. Keywords: schizophrenia; GWAS; causal genetic variants; enhancers; brain epigenomics; genome/epigenome editing 1. Introduction Schizophrenia is a severe mental illness that affects between 0.5% and 0.7% of the human population [1]. Both environmental and genetic factors are thought to be involved in its pathogenesis, with genetic factors playing a key role in disease risk, as the heritability of schizophrenia is estimated to be 70–85% [2,3]. -

Research Article a Robust Topology-Based Algorithm for Gene Expression Profiling
International Scholarly Research Network ISRN Bioinformatics Volume 2012, Article ID 381023, 11 pages doi:10.5402/2012/381023 Research Article A Robust Topology-Based Algorithm for Gene Expression Profiling Lars Seemann,1 Jason Shulman,2 and Gemunu H. Gunaratne1 1 Department of Physics, University of Houston, Houston, TX 77204, USA 2 Department of Physics, Richard Stockton College of New Jersey, Pomona, NJ 08240, USA Correspondence should be addressed to Gemunu H. Gunaratne, [email protected] Received 3 August 2012; Accepted 30 August 2012 Academic Editors: B. Haubold and K. Yura Copyright © 2012 Lars Seemann et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Early and accurate diagnoses of cancer can significantly improve the design of personalized therapy and enhance the success of therapeutic interventions. Histopathological approaches, which rely on microscopic examinations of malignant tissue, are not conducive to timely diagnoses. High throughput genomics offers a possible new classification of cancer subtypes. Unfortunately, most clustering algorithms have not been proven sufficiently robust. We propose a novel approach that relies on the use of statistical invariants and persistent homology, one of the most exciting recent developments in topology. It identifies a sufficient but compact set of genes for the analysis as well as a core group of tightly correlated patient samples for each subtype. Partitioning occurs hierarchically and allows for the identification of genetically similar subtypes. We analyzed the gene expression profiles of 202 tumors of the brain cancer glioblastoma multiforme (GBM) given at the Cancer Genome Atlas (TCGA) site. -

In This Table Protein Name, Uniprot Code, Gene Name P-Value
Supplementary Table S1: In this table protein name, uniprot code, gene name p-value and Fold change (FC) for each comparison are shown, for 299 of the 301 significantly regulated proteins found in both comparisons (p-value<0.01, fold change (FC) >+/-0.37) ALS versus control and FTLD-U versus control. Two uncharacterized proteins have been excluded from this list Protein name Uniprot Gene name p value FC FTLD-U p value FC ALS FTLD-U ALS Cytochrome b-c1 complex P14927 UQCRB 1.534E-03 -1.591E+00 6.005E-04 -1.639E+00 subunit 7 NADH dehydrogenase O95182 NDUFA7 4.127E-04 -9.471E-01 3.467E-05 -1.643E+00 [ubiquinone] 1 alpha subcomplex subunit 7 NADH dehydrogenase O43678 NDUFA2 3.230E-04 -9.145E-01 2.113E-04 -1.450E+00 [ubiquinone] 1 alpha subcomplex subunit 2 NADH dehydrogenase O43920 NDUFS5 1.769E-04 -8.829E-01 3.235E-05 -1.007E+00 [ubiquinone] iron-sulfur protein 5 ARF GTPase-activating A0A0C4DGN6 GIT1 1.306E-03 -8.810E-01 1.115E-03 -7.228E-01 protein GIT1 Methylglutaconyl-CoA Q13825 AUH 6.097E-04 -7.666E-01 5.619E-06 -1.178E+00 hydratase, mitochondrial ADP/ATP translocase 1 P12235 SLC25A4 6.068E-03 -6.095E-01 3.595E-04 -1.011E+00 MIC J3QTA6 CHCHD6 1.090E-04 -5.913E-01 2.124E-03 -5.948E-01 MIC J3QTA6 CHCHD6 1.090E-04 -5.913E-01 2.124E-03 -5.948E-01 Protein kinase C and casein Q9BY11 PACSIN1 3.837E-03 -5.863E-01 3.680E-06 -1.824E+00 kinase substrate in neurons protein 1 Tubulin polymerization- O94811 TPPP 6.466E-03 -5.755E-01 6.943E-06 -1.169E+00 promoting protein MIC C9JRZ6 CHCHD3 2.912E-02 -6.187E-01 2.195E-03 -9.781E-01 Mitochondrial 2- -
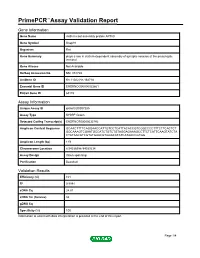
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name clathrin coat assembly protein AP180 Gene Symbol Snap91 Organism Rat Gene Summary plays a role in clathrin-dependent assembly of synaptic vesicles at the presynaptic terminal Gene Aliases Not Available RefSeq Accession No. NM_031728 UniGene ID Rn.11022,Rn.164718 Ensembl Gene ID ENSRNOG00000023861 Entrez Gene ID 65178 Assay Information Unique Assay ID qRnoCID0007285 Assay Type SYBR® Green Detected Coding Transcript(s) ENSRNOT00000032792 Amplicon Context Sequence GCAACTTTTCAGGAACCATTGTCCTCATTACACCGTCGGCCCCTTTCTTCACTCT GGCAAAGTCGAATGCCATCTGTCTGTAGGAGAAAGCCTTCTCATTCAAGTATCTA CTATAACGTCGTATGAACGTGGACATATCATAACCGTGG Amplicon Length (bp) 119 Chromosome Location 8:94036996-94039234 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 101 R2 0.9991 cDNA Cq 24.81 cDNA Tm (Celsius) 82 gDNA Cq Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report Snap91, Rat Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. Detected Coding Transcript(s) This is a list of the Ensembl transcript ID(s) that this assay will detect. -

Effects of Chronic Stress on Prefrontal Cortex Transcriptome in Mice Displaying Different Genetic Backgrounds
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Springer - Publisher Connector J Mol Neurosci (2013) 50:33–57 DOI 10.1007/s12031-012-9850-1 Effects of Chronic Stress on Prefrontal Cortex Transcriptome in Mice Displaying Different Genetic Backgrounds Pawel Lisowski & Marek Wieczorek & Joanna Goscik & Grzegorz R. Juszczak & Adrian M. Stankiewicz & Lech Zwierzchowski & Artur H. Swiergiel Received: 14 May 2012 /Accepted: 25 June 2012 /Published online: 27 July 2012 # The Author(s) 2012. This article is published with open access at Springerlink.com Abstract There is increasing evidence that depression signaling pathway (Clic6, Drd1a,andPpp1r1b). LA derives from the impact of environmental pressure on transcriptome affected by CMS was associated with genetically susceptible individuals. We analyzed the genes involved in behavioral response to stimulus effects of chronic mild stress (CMS) on prefrontal cor- (Fcer1g, Rasd2, S100a8, S100a9, Crhr1, Grm5,and tex transcriptome of two strains of mice bred for high Prkcc), immune effector processes (Fcer1g, Mpo,and (HA)and low (LA) swim stress-induced analgesia that Igh-VJ558), diacylglycerol binding (Rasgrp1, Dgke, differ in basal transcriptomic profiles and depression- Dgkg,andPrkcc), and long-term depression (Crhr1, like behaviors. We found that CMS affected 96 and 92 Grm5,andPrkcc) and/or coding elements of dendrites genes in HA and LA mice, respectively. Among genes (Crmp1, Cntnap4,andPrkcc) and myelin proteins with the same expression pattern in both strains after (Gpm6a, Mal,andMog). The results indicate significant CMS, we observed robust upregulation of Ttr gene contribution of genetic background to differences in coding transthyretin involved in amyloidosis, seizures, stress response gene expression in the mouse prefrontal stroke-like episodes, or dementia. -

Correlated Expression Analysis of Genes Implicated in Schizophrenia
Author’s Accepted Manuscript Correlated expression analysis of genes implicated in schizophrenia: identification of putative disease- related pathways Erin I. Liedtke, Sirey Zhang, John A. Thompson, Stefan Sillau, Judith Gault www.elsevier.com/locate/nhtm PII: S2307-5023(16)30043-1 DOI: http://dx.doi.org/10.1016/j.nhtm.2016.11.002 Reference: NHTM39 To appear in: New Horizons in Translational Medicine Received date: 27 October 2016 Revised date: 23 November 2016 Accepted date: 30 November 2016 Cite this article as: Erin I. Liedtke, Sirey Zhang, John A. Thompson, Stefan Sillau and Judith Gault, Correlated expression analysis of genes implicated in schizophrenia: identification of putative disease-related pathways, New Horizons in Translational Medicine, http://dx.doi.org/10.1016/j.nhtm.2016.11.002 This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting galley proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain. Correlated expression analysis of genes implicated in schizophrenia: identification of putative disease-related pathways Erin I Liedtke1a, Sirey Zhanga1, John A. Thompson, PhDa, Stefan Sillau, PhDb, Judith Gault, PhDa,c* aUniversity of Colorado Denver, Anschutz Medical Campus, Department of Neurosurgery bUniversity of Colorado Denver, Anschutz Medical Campus, Department of Neurology cUniversity of Colorado Denver, Anschutz Medical Campus, Department of Psychiatry *Contact: Judith Gault, PhD 12700 E. -

WO 2016/040794 Al 17 March 2016 (17.03.2016) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/040794 Al 17 March 2016 (17.03.2016) P O P C T (51) International Patent Classification: AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, C12N 1/19 (2006.01) C12Q 1/02 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, C12N 15/81 (2006.01) C07K 14/47 (2006.01) DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, (21) International Application Number: KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, PCT/US20 15/049674 MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (22) International Filing Date: PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, 11 September 2015 ( 11.09.201 5) SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every (26) Publication Language: English kind of regional protection available): ARIPO (BW, GH, (30) Priority Data: GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, 62/050,045 12 September 2014 (12.09.2014) US TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, (71) Applicant: WHITEHEAD INSTITUTE FOR BIOMED¬ DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, ICAL RESEARCH [US/US]; Nine Cambridge Center, LV, MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, Cambridge, Massachusetts 02142-1479 (US). -

PLEK2, RRM2, GCSH: a Novel WWOX-Dependent Biomarker Triad of Glioblastoma at the Crossroads of Cytoskeleton Reorganization and Metabolism Alterations
cancers Article PLEK2, RRM2, GCSH: A Novel WWOX-Dependent Biomarker Triad of Glioblastoma at the Crossroads of Cytoskeleton Reorganization and Metabolism Alterations Zaneta˙ Kałuzi ´nska* , Damian Kołat , Andrzej K. Bednarek and Elzbieta˙ Płuciennik Department of Molecular Carcinogenesis, Medical University of Lodz, 90-752 Lodz, Poland; [email protected] (D.K.); [email protected] (A.K.B.); [email protected] (E.P.) * Correspondence: [email protected] Simple Summary: Cytoskeleton reorganization affects the malignancy of glioblastoma. The WWOX gene is a tumor suppressor in glioblastoma and was found to modulate the cytoskeletal machinery in neural progenitor cells. To date, the role of this gene in the cytoskeleton or glioblastoma has been studied separately. Therefore, the purpose of this study was to investigate WWOX-dependent genes in glioblastoma and indicate cytoskeleton-related processes they are involved in. The most relevant WWOX-dependent genes were found to be PLEK2, RRM2, and GCSH, which have been proposed as novel biomarkers. Their biological functions suggest that there is an important link between cytoskeleton and metabolism, orchestrating tumor proliferation, metastasis, and resistance. Searching for such new therapeutic targets is important due to the constant lack of effective treatment ˙ Citation: Kałuzi´nska, Z.; Kołat, D.; for glioblastoma patients. Bednarek, A.K.; Płuciennik, E. PLEK2, RRM2, GCSH: A Novel Abstract: Glioblastoma is one of the deadliest human cancers. Its malignancy depends on cytoskele- WWOX-Dependent Biomarker Triad ton reorganization, which is related to, e.g., epithelial-to-mesenchymal transition and metastasis. The of Glioblastoma at the Crossroads of malignant phenotype of glioblastoma is also affected by the WWOX gene, which is lost in nearly Cytoskeleton Reorganization and a quarter of gliomas. -

Supplementary Table 3. Genes Specifically Regulated by Zol (Non-Significant for Fluva)
Supplementary Table 3. Genes specifically regulated by Zol (non-significant for Fluva). log2 Genes Probe Genes Symbol Genes Title Zol100 vs Zol vs Set ID Control (24h) Control (48h) 8065412 CST1 cystatin SN 2,168 1,772 7928308 DDIT4 DNA-damage-inducible transcript 4 2,066 0,349 8154100 VLDLR very low density lipoprotein 1,99 0,413 receptor 8149749 TNFRSF10D tumor necrosis factor receptor 1,973 0,659 superfamily, member 10d, decoy with truncated death domain 8006531 SLFN5 schlafen family member 5 1,692 0,183 8147145 ATP6V0D2 ATPase, H+ transporting, lysosomal 1,689 0,71 38kDa, V0 subunit d2 8013660 ALDOC aldolase C, fructose-bisphosphate 1,649 0,871 8140967 SAMD9 sterile alpha motif domain 1,611 0,66 containing 9 8113709 LOX lysyl oxidase 1,566 0,524 7934278 P4HA1 prolyl 4-hydroxylase, alpha 1,527 0,428 polypeptide I 8027002 GDF15 growth differentiation factor 15 1,415 0,201 7961175 KLRC3 killer cell lectin-like receptor 1,403 1,038 subfamily C, member 3 8081288 TMEM45A transmembrane protein 45A 1,342 0,401 8012126 CLDN7 claudin 7 1,339 0,415 7993588 TMC7 transmembrane channel-like 7 1,318 0,3 8073088 APOBEC3G apolipoprotein B mRNA editing 1,302 0,174 enzyme, catalytic polypeptide-like 3G 8046408 PDK1 pyruvate dehydrogenase kinase, 1,287 0,382 isozyme 1 8161174 GNE glucosamine (UDP-N-acetyl)-2- 1,283 0,562 epimerase/N-acetylmannosamine kinase 7937079 BNIP3 BCL2/adenovirus E1B 19kDa 1,278 0,5 interacting protein 3 8043283 KDM3A lysine (K)-specific demethylase 3A 1,274 0,453 7923991 PLXNA2 plexin A2 1,252 0,481 8163618 TNFSF15 tumor necrosis -

Comparative Gene Expression Analysis Reveals a Characteristic Molecular Profile of the Superior Olivary Complex
tapraid5/z3x-anrec/z3x-anrec/z3x00406/z3x1168d06g royerl Sϭ7 3/1/06 4:24 Art: 05-0227 THE ANATOMICAL RECORD PART A 00A:000–000 (2006) Comparative Gene Expression Analysis Reveals a Characteristic Molecular Profile of the Superior Olivary Complex HANS GERD NOTHWANG,* ALEXANDER KOEHL, AND ECKHARD FRIAUF Abteilung Tierphysiologie, Technische Universita¨t Kaiserslautern, Kaiserslautern, Germany ABSTRACT The superior olivary complex (SOC) is a very conspicuous structure in the mammalian auditory brainstem. It represents the first binaural processing center and is important for sound localization in the azimuth and in feedback regulation of cochlear function. In order to define molecular determinants of the SOC, which are of potential functional relevance, we have performed a comprehensive analysis of its transcriptome by serial analysis of gene expression in adult rats. Here, we performed a detailed analysis of the SOC’s gene expression profile compared to that of two other neural tissues, the striatum and the hippocampus, and with extraocular muscle tissue. This tested the hypothesis that SOC-specific or significantly upregulated transcripts provide candidates for the specific function of auditory neurons. Thirty-three genes were significantly upregulated in the SOC when compared to the two other neural tissues. Thirteen encoded proteins involved in neurotransmission, including action potential propagation, exocytosis, and myelination; five genes are important for the energy metabolism; and five transcripts are unknown or poorly characterized and have yet to be described in the nervous system. The comparison of functional gene classes indicates that the SOC has the highest energy demand of the three neural tissues, yet protein turnover is apparently not increased. -

How Alternative Splicing Affects Membrane-Trafficking Dynamics R
© 2018. Published by The Company of Biologists Ltd | Journal of Cell Science (2018) 131, jcs216465. doi:10.1242/jcs.216465 REVIEW How alternative splicing affects membrane-trafficking dynamics R. Eric Blue1, Ennessa G. Curry1,*, Nichlas M. Engels1,*, Eunice Y. Lee1 and Jimena Giudice1,2,3,‡ ABSTRACT et al., 2012). Tissue-specific exons encode disordered segments The cell biology field has outstanding working knowledge of the within proteins that function in microtubule-based transport, fundamentals of membrane-trafficking pathways, which are of critical endocytosis and membrane deformation (Buljan et al., 2012; Ellis importance in health and disease. Current challenges include et al., 2012) (Box 1). understanding how trafficking pathways are fine-tuned for In humans, 90-95% of genes undergo alternative splicing, specialized tissue functions in vivo and during development. In expanding protein function beyond genetic diversity (Pan et al., parallel, the ENCODE project and numerous genetic studies have 2008; Wang et al., 2008). Splicing of intronic regions is regulated by revealed that alternative splicing regulates gene expression in tissues the strength of the splice sites; strong splice sites lead to constitutive and throughout development at a post-transcriptional level. This splicing, whereas weak splice sites are used in a context-dependent Review summarizes recent discoveries demonstrating that alternative manner (alternative splicing). Usage of weak splice sites is regulated splicing affects tissue specialization and membrane-trafficking by cis-regulatory sequences, trans-acting factors such as RNA- proteins during development, and examines how this regulation is binding proteins (RBPs) and epigenetics (Kornblihtt et al., 2013). altered in human disease. We first discuss how alternative splicing of Depending on splice site locations, different types of alternative clathrin, SNAREs and BAR-domain proteins influences endocytosis, splicing events are produced, which comprise insertion of secretion and membrane dynamics, respectively. -

Deletion 6Q Drives T-Cell Leukemia Progression by Ribosome Modulation
Published OnlineFirst September 28, 2018; DOI: 10.1158/2159-8290.CD-17-0831 RESEARCH ARTICLE Deletion 6q Drives T-cell Leukemia Progression by Ribosome Modulation Stéphanie Gachet 1 , 2 , Tiama El-Chaar 1 , 2 , David Avran 1 , 2 , 3 , Eulalia Genesca 1 , 2 , Frédéric Catez 4 , Samuel Quentin1 , 2 , 3 , Marc Delord 2 , Gabriel Thérizols 4 , Delphine Briot 1 , 2 , 3 , Godelieve Meunier 1 , 2 , Lucie Hernandez1 , 2 , Marika Pla 2 , 5 , Willem K. Smits 6 , Jessica G. Buijs-Gladdines 6 , Wouter Van Loocke 7 , Gerben Menschaert7 , Isabelle André-Schmutz 8 , Tom Taghon 7 , Pieter Van Vlierberghe 7 , Jules P. Meijerink 6 , André Baruchel2 , 9 , Hervé Dombret 2 , 10 , Emmanuelle Clappier 1 , 2 , 3 , Jean-Jacques Diaz 4 , Claude Gazin 11 , Hugues de Thé1 , 2 , François Sigaux 1 , 2 , 3 , and Jean Soulier 1 , 2 , 3 Downloaded from cancerdiscovery.aacrjournals.org on September 27, 2021. © 2018 American Association for Cancer Research. Published OnlineFirst September 28, 2018; DOI: 10.1158/2159-8290.CD-17-0831 ABSTRACT Deletion of chromosome 6q is a well-recognized abnormality found in poor- prognosis T-cell acute lymphoblastic leukemia (T-ALL). Using integrated genomic approaches, we identifi ed two candidate haploinsuffi cient genes contiguous at 6q14,SYNCRIP (encod- ing hnRNP-Q) and SNHG5 (that hosts snoRNAs), both involved in regulating RNA maturation and trans- lation. Combined silencing of both genes, but not of either gene alone, accelerated leukemogeneis in a Tal1/Lmo1/Notch1 -driven mouse model, demonstrating the tumor-suppressive nature of the two-gene region. Proteomic and translational profi ling of cells in which we engineered a short 6q deletion by CRISPR/Cas9 genome editing indicated decreased ribosome and mitochondrial activities, suggesting that the resulting metabolic changes may regulate tumor progression.