Deletion 6Q Drives T-Cell Leukemia Progression by Ribosome Modulation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Downloads/ (Accessed on 17 January 2020)
cells Review Novel Approaches for Identifying the Molecular Background of Schizophrenia Arkadiy K. Golov 1,2,*, Nikolay V. Kondratyev 1 , George P. Kostyuk 3 and Vera E. Golimbet 1 1 Mental Health Research Center, 34 Kashirskoye shosse, 115522 Moscow, Russian; [email protected] (N.V.K.); [email protected] (V.E.G.) 2 Institute of Gene Biology, Russian Academy of Sciences, 34/5 Vavilova Street, 119334 Moscow, Russian 3 Alekseev Psychiatric Clinical Hospital No. 1, 2 Zagorodnoye shosse, 115191 Moscow, Russian; [email protected] * Correspondence: [email protected] Received: 5 November 2019; Accepted: 16 January 2020; Published: 18 January 2020 Abstract: Recent advances in psychiatric genetics have led to the discovery of dozens of genomic loci associated with schizophrenia. However, a gap exists between the detection of genetic associations and understanding the underlying molecular mechanisms. This review describes the basic approaches used in the so-called post-GWAS studies to generate biological interpretation of the existing population genetic data, including both molecular (creation and analysis of knockout animals, exploration of the transcriptional effects of common variants in human brain cells) and computational (fine-mapping of causal variability, gene set enrichment analysis, partitioned heritability analysis) methods. The results of the crucial studies, in which these approaches were used to uncover the molecular and neurobiological basis of the disease, are also reported. Keywords: schizophrenia; GWAS; causal genetic variants; enhancers; brain epigenomics; genome/epigenome editing 1. Introduction Schizophrenia is a severe mental illness that affects between 0.5% and 0.7% of the human population [1]. Both environmental and genetic factors are thought to be involved in its pathogenesis, with genetic factors playing a key role in disease risk, as the heritability of schizophrenia is estimated to be 70–85% [2,3]. -

Datasheet: VPA00486 Product Details
Datasheet: VPA00486 Description: RABBIT ANTI hnRNPQ / SYNCRIP Specificity: hnRNP Q / SYNCRIP Format: Semi Pure Product Type: PrecisionAb™ Polyclonal Isotype: Polyclonal IgG Quantity: 100 µl Product Details Applications This product has been reported to work in the following applications. This information is derived from testing within our laboratories, peer-reviewed publications or personal communications from the originators. Please refer to references indicated for further information. For general protocol recommendations, please visit www.bio-rad-antibodies.com/protocols. Yes No Not Determined Suggested Dilution Western Blotting 1/1000 PrecisionAb antibodies have been extensively validated for the western blot application. The antibody has been validated at the suggested dilution. Where this product has not been tested for use in a particular technique this does not necessarily exclude its use in such procedures. Further optimization may be required dependant on sample type. Target Species Human Species Cross Reacts with: Mouse Reactivity N.B. Antibody reactivity and working conditions may vary between species. Product Form Purified IgG - liquid Preparation Rabbit Ig fraction prepared by ammonium sulphate precipitation Buffer Solution Phosphate buffered saline Preservative 0.09% Sodium Azide (NaN ) Stabilisers 3 Immunogen KLH conjugated synthetic peptide corresponding to amino acid 591-623 of human hnRNP Q External Database Links UniProt: O60506 Related reagents Entrez Gene: 10492 SYNCRIP Related reagents Synonyms HNRPQ, NSAP1 Page 1 of 2 Specificity Rabbit anti Human hnRNP Q / SYNCRIP antibody recognizes hnRNP Q / SYNCRIP also known as heterogeneous nuclear ribonucleoprotein Q, NS1-associated protein 1, glycine- and tyrosine-rich RNA-binding protein or Synaptotagmin-binding, cytoplasmic RNA-interacting protein. Encode by the SYNCRIP gene, hnRNP Q is a member of the cellular heterogeneous nuclear ribonucleoprotein (hnRNP) family. -

Posttranscriptional Regulation of HLA-A Protein Expression By
Posttranscriptional Regulation of HLA-A Protein Expression by Alternative Polyadenylation Signals Involving the RNA-Binding Protein Syncrip This information is current as of September 27, 2021. Smita Kulkarni, Veron Ramsuran, Marijana Rucevic, Sukhvinder Singh, Alexandra Lied, Viraj Kulkarni, Colm O'hUigin, Sylvie Le Gall and Mary Carrington J Immunol 2017; 199:3892-3899; Prepublished online 20 October 2017; Downloaded from doi: 10.4049/jimmunol.1700697 http://www.jimmunol.org/content/199/11/3892 http://www.jimmunol.org/ Supplementary http://www.jimmunol.org/content/suppl/2017/10/20/jimmunol.170069 Material 7.DCSupplemental References This article cites 66 articles, 30 of which you can access for free at: http://www.jimmunol.org/content/199/11/3892.full#ref-list-1 Why The JI? Submit online. by guest on September 27, 2021 • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2017 by The American -

Network-Based Prediction of Polygenic Disease Genes Involved in Cell Motility Miriam Bern†, Alexander King†, Derek A
Bern et al. BMC Bioinformatics 2019, 20(Suppl 12):313 https://doi.org/10.1186/s12859-019-2834-1 RESEARCH Open Access Network-based prediction of polygenic disease genes involved in cell motility Miriam Bern†, Alexander King†, Derek A. Applewhite and Anna Ritz* From International Workshop on Computational Network Biology: Modeling, Analysis and Control Washington, D.C., USA. 29 August 2018 Abstract Background: Schizophrenia and autism are examples of polygenic diseases caused by a multitude of genetic variants, many of which are still poorly understood. Recently, both diseases have been associated with disrupted neuron motility and migration patterns, suggesting that aberrant cell motility is a phenotype for these neurological diseases. Results: We formulate the POLYGENIC DISEASE PHENOTYPE Problem which seeks to identify candidate disease genes that may be associated with a phenotype such as cell motility. We present a machine learning approach to solve this problem for schizophrenia and autism genes within a brain-specific functional interaction network. Our method outperforms peer semi-supervised learning approaches, achieving better cross-validation accuracy across different sets of gold-standard positives. We identify top candidates for both schizophrenia and autism, and select six genes labeled as schizophrenia positives that are predicted to be associated with cell motility for follow-up experiments. Conclusions: Candidate genes predicted by our method suggest testable hypotheses about these genes’ role in cell motility regulation, offering a framework for generating predictions for experimental validation. Keywords: Semi-supervised learning, Functional interaction network, Schizophrenia, Autism, Cell motility Background participants [4, 5], indicating that schizophrenia arises Many polygenic diseases, which arise from the action from a multitude of cellular perturbations compounding or influence of multiple genes, are difficult to geneti- their effects. -

Research Article a Robust Topology-Based Algorithm for Gene Expression Profiling
International Scholarly Research Network ISRN Bioinformatics Volume 2012, Article ID 381023, 11 pages doi:10.5402/2012/381023 Research Article A Robust Topology-Based Algorithm for Gene Expression Profiling Lars Seemann,1 Jason Shulman,2 and Gemunu H. Gunaratne1 1 Department of Physics, University of Houston, Houston, TX 77204, USA 2 Department of Physics, Richard Stockton College of New Jersey, Pomona, NJ 08240, USA Correspondence should be addressed to Gemunu H. Gunaratne, [email protected] Received 3 August 2012; Accepted 30 August 2012 Academic Editors: B. Haubold and K. Yura Copyright © 2012 Lars Seemann et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Early and accurate diagnoses of cancer can significantly improve the design of personalized therapy and enhance the success of therapeutic interventions. Histopathological approaches, which rely on microscopic examinations of malignant tissue, are not conducive to timely diagnoses. High throughput genomics offers a possible new classification of cancer subtypes. Unfortunately, most clustering algorithms have not been proven sufficiently robust. We propose a novel approach that relies on the use of statistical invariants and persistent homology, one of the most exciting recent developments in topology. It identifies a sufficient but compact set of genes for the analysis as well as a core group of tightly correlated patient samples for each subtype. Partitioning occurs hierarchically and allows for the identification of genetically similar subtypes. We analyzed the gene expression profiles of 202 tumors of the brain cancer glioblastoma multiforme (GBM) given at the Cancer Genome Atlas (TCGA) site. -

In This Table Protein Name, Uniprot Code, Gene Name P-Value
Supplementary Table S1: In this table protein name, uniprot code, gene name p-value and Fold change (FC) for each comparison are shown, for 299 of the 301 significantly regulated proteins found in both comparisons (p-value<0.01, fold change (FC) >+/-0.37) ALS versus control and FTLD-U versus control. Two uncharacterized proteins have been excluded from this list Protein name Uniprot Gene name p value FC FTLD-U p value FC ALS FTLD-U ALS Cytochrome b-c1 complex P14927 UQCRB 1.534E-03 -1.591E+00 6.005E-04 -1.639E+00 subunit 7 NADH dehydrogenase O95182 NDUFA7 4.127E-04 -9.471E-01 3.467E-05 -1.643E+00 [ubiquinone] 1 alpha subcomplex subunit 7 NADH dehydrogenase O43678 NDUFA2 3.230E-04 -9.145E-01 2.113E-04 -1.450E+00 [ubiquinone] 1 alpha subcomplex subunit 2 NADH dehydrogenase O43920 NDUFS5 1.769E-04 -8.829E-01 3.235E-05 -1.007E+00 [ubiquinone] iron-sulfur protein 5 ARF GTPase-activating A0A0C4DGN6 GIT1 1.306E-03 -8.810E-01 1.115E-03 -7.228E-01 protein GIT1 Methylglutaconyl-CoA Q13825 AUH 6.097E-04 -7.666E-01 5.619E-06 -1.178E+00 hydratase, mitochondrial ADP/ATP translocase 1 P12235 SLC25A4 6.068E-03 -6.095E-01 3.595E-04 -1.011E+00 MIC J3QTA6 CHCHD6 1.090E-04 -5.913E-01 2.124E-03 -5.948E-01 MIC J3QTA6 CHCHD6 1.090E-04 -5.913E-01 2.124E-03 -5.948E-01 Protein kinase C and casein Q9BY11 PACSIN1 3.837E-03 -5.863E-01 3.680E-06 -1.824E+00 kinase substrate in neurons protein 1 Tubulin polymerization- O94811 TPPP 6.466E-03 -5.755E-01 6.943E-06 -1.169E+00 promoting protein MIC C9JRZ6 CHCHD3 2.912E-02 -6.187E-01 2.195E-03 -9.781E-01 Mitochondrial 2- -
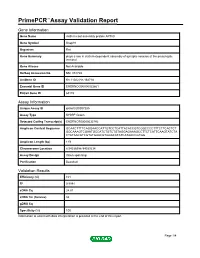
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name clathrin coat assembly protein AP180 Gene Symbol Snap91 Organism Rat Gene Summary plays a role in clathrin-dependent assembly of synaptic vesicles at the presynaptic terminal Gene Aliases Not Available RefSeq Accession No. NM_031728 UniGene ID Rn.11022,Rn.164718 Ensembl Gene ID ENSRNOG00000023861 Entrez Gene ID 65178 Assay Information Unique Assay ID qRnoCID0007285 Assay Type SYBR® Green Detected Coding Transcript(s) ENSRNOT00000032792 Amplicon Context Sequence GCAACTTTTCAGGAACCATTGTCCTCATTACACCGTCGGCCCCTTTCTTCACTCT GGCAAAGTCGAATGCCATCTGTCTGTAGGAGAAAGCCTTCTCATTCAAGTATCTA CTATAACGTCGTATGAACGTGGACATATCATAACCGTGG Amplicon Length (bp) 119 Chromosome Location 8:94036996-94039234 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 101 R2 0.9991 cDNA Cq 24.81 cDNA Tm (Celsius) 82 gDNA Cq Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report Snap91, Rat Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. Detected Coding Transcript(s) This is a list of the Ensembl transcript ID(s) that this assay will detect. -

Effects of Chronic Stress on Prefrontal Cortex Transcriptome in Mice Displaying Different Genetic Backgrounds
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Springer - Publisher Connector J Mol Neurosci (2013) 50:33–57 DOI 10.1007/s12031-012-9850-1 Effects of Chronic Stress on Prefrontal Cortex Transcriptome in Mice Displaying Different Genetic Backgrounds Pawel Lisowski & Marek Wieczorek & Joanna Goscik & Grzegorz R. Juszczak & Adrian M. Stankiewicz & Lech Zwierzchowski & Artur H. Swiergiel Received: 14 May 2012 /Accepted: 25 June 2012 /Published online: 27 July 2012 # The Author(s) 2012. This article is published with open access at Springerlink.com Abstract There is increasing evidence that depression signaling pathway (Clic6, Drd1a,andPpp1r1b). LA derives from the impact of environmental pressure on transcriptome affected by CMS was associated with genetically susceptible individuals. We analyzed the genes involved in behavioral response to stimulus effects of chronic mild stress (CMS) on prefrontal cor- (Fcer1g, Rasd2, S100a8, S100a9, Crhr1, Grm5,and tex transcriptome of two strains of mice bred for high Prkcc), immune effector processes (Fcer1g, Mpo,and (HA)and low (LA) swim stress-induced analgesia that Igh-VJ558), diacylglycerol binding (Rasgrp1, Dgke, differ in basal transcriptomic profiles and depression- Dgkg,andPrkcc), and long-term depression (Crhr1, like behaviors. We found that CMS affected 96 and 92 Grm5,andPrkcc) and/or coding elements of dendrites genes in HA and LA mice, respectively. Among genes (Crmp1, Cntnap4,andPrkcc) and myelin proteins with the same expression pattern in both strains after (Gpm6a, Mal,andMog). The results indicate significant CMS, we observed robust upregulation of Ttr gene contribution of genetic background to differences in coding transthyretin involved in amyloidosis, seizures, stress response gene expression in the mouse prefrontal stroke-like episodes, or dementia. -

Large-Scale Analysis of Genome and Transcriptome Alterations in Multiple Tumors Unveils Novel Cancer-Relevant Splicing Networks
Downloaded from genome.cshlp.org on October 2, 2021 - Published by Cold Spring Harbor Laboratory Press Large-scale analysis of genome and transcriptome alterations in multiple tumors unveils novel cancer-relevant splicing networks Endre Sebestyén1,*, Babita Singh1,*, Belén Miñana1,2, Amadís Pagès1, Francesca Mateo3, Miguel Angel Pujana3, Juan Valcárcel1,2,4, Eduardo Eyras1,4,5 1Universitat Pompeu Fabra, Dr. Aiguader 88, E08003 Barcelona, Spain 2Centre for Genomic Regulation, Dr. Aiguader 88, E08003 Barcelona, Spain 3Program Against Cancer Therapeutic Resistance (ProCURE), Catalan Institute of Oncology (ICO), Bellvitge Institute for Biomedical Research (IDIBELL), E08908 L’Hospitalet del Llobregat, Spain. 4Catalan Institution for Research and Advanced Studies, Passeig Lluís Companys 23, E08010 Barcelona, Spain *Equal contribution 5Correspondence to: [email protected] Keywords: alternative splicing, RNA binding proteins, splicing networks, cancer 1 Downloaded from genome.cshlp.org on October 2, 2021 - Published by Cold Spring Harbor Laboratory Press Abstract Alternative splicing is regulated by multiple RNA-binding proteins and influences the expression of most eukaryotic genes. However, the role of this process in human disease, and particularly in cancer, is only starting to be unveiled. We systematically analyzed mutation, copy number and gene expression patterns of 1348 RNA-binding protein (RBP) genes in 11 solid tumor types, together with alternative splicing changes in these tumors and the enrichment of binding motifs in the alternatively spliced sequences. Our comprehensive study reveals widespread alterations in the expression of RBP genes, as well as novel mutations and copy number variations in association with multiple alternative splicing changes in cancer drivers and oncogenic pathways. Remarkably, the altered splicing patterns in several tumor types recapitulate those of undifferentiated cells. -

Correlated Expression Analysis of Genes Implicated in Schizophrenia
Author’s Accepted Manuscript Correlated expression analysis of genes implicated in schizophrenia: identification of putative disease- related pathways Erin I. Liedtke, Sirey Zhang, John A. Thompson, Stefan Sillau, Judith Gault www.elsevier.com/locate/nhtm PII: S2307-5023(16)30043-1 DOI: http://dx.doi.org/10.1016/j.nhtm.2016.11.002 Reference: NHTM39 To appear in: New Horizons in Translational Medicine Received date: 27 October 2016 Revised date: 23 November 2016 Accepted date: 30 November 2016 Cite this article as: Erin I. Liedtke, Sirey Zhang, John A. Thompson, Stefan Sillau and Judith Gault, Correlated expression analysis of genes implicated in schizophrenia: identification of putative disease-related pathways, New Horizons in Translational Medicine, http://dx.doi.org/10.1016/j.nhtm.2016.11.002 This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting galley proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain. Correlated expression analysis of genes implicated in schizophrenia: identification of putative disease-related pathways Erin I Liedtke1a, Sirey Zhanga1, John A. Thompson, PhDa, Stefan Sillau, PhDb, Judith Gault, PhDa,c* aUniversity of Colorado Denver, Anschutz Medical Campus, Department of Neurosurgery bUniversity of Colorado Denver, Anschutz Medical Campus, Department of Neurology cUniversity of Colorado Denver, Anschutz Medical Campus, Department of Psychiatry *Contact: Judith Gault, PhD 12700 E. -

SYNCRIP Facilitates Porcine Parvovirus Viral DNA Replication
Chen et al. Vet Res (2021) 52:73 https://doi.org/10.1186/s13567-021-00938-6 RESEARCH ARTICLE Open Access SYNCRIP facilitates porcine parvovirus viral DNA replication through the alternative splicing of NS1 mRNA to promote NS2 mRNA formation Songbiao Chen†, Bichen Miao†, Nannan Chen, Caiyi Chen, Ting Shao, Xuezhi Zhang, Lingling Chang, Xiujuan Zhang, Qian Du, Yong Huang* and Dewen Tong* Abstract Porcine Parvovirus (PPV), a pathogen causing porcine reproductive disorders, encodes two capsid proteins (VP1 and VP2) and three nonstructural proteins (NS1, NS2 and SAT) in infected cells. The PPV NS2 mRNA is from NS1 mRNA after alternative splicing, yet the corresponding mechanism is unclear. In this study, we identifed a PPV NS1 mRNA bind- ing protein SYNCRIP, which belongs to the hnRNP family and has been identifed to be involved in host pre-mRNA splicing by RNA-pulldown and mass spectrometry approaches. SYNCRIP was found to be signifcantly up-regulated by PPV infection in vivo and in vitro. We confrmed that it directly interacts with PPV NS1 mRNA and is co-localized at the cytoplasm in PPV-infected cells. Overexpression of SYNCRIP signifcantly reduced the NS1 mRNA and protein levels, whereas deletion of SYNCRIP signifcantly reduced NS2 mRNA and protein levels and the ratio of NS2 to NS1, and further impaired replication of the PPV. Furthermore, we found that SYNCRIP was able to bind the 3′-terminal site of NS1 mRNA to promote the cleavage of NS1 mRNA into NS2 mRNA. Taken together, the results presented here dem- onstrate that SYNCRIP is a critical molecule in the alternative splicing process of PPV mRNA, while revealing a novel function for this protein and providing a potential target of antiviral intervention for the control of porcine parvovirus disease.