Research Article a Robust Topology-Based Algorithm for Gene Expression Profiling
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Transcriptional Control of Tissue-Resident Memory T Cell Generation
Transcriptional control of tissue-resident memory T cell generation Filip Cvetkovski Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2019 © 2019 Filip Cvetkovski All rights reserved ABSTRACT Transcriptional control of tissue-resident memory T cell generation Filip Cvetkovski Tissue-resident memory T cells (TRM) are a non-circulating subset of memory that are maintained at sites of pathogen entry and mediate optimal protection against reinfection. Lung TRM can be generated in response to respiratory infection or vaccination, however, the molecular pathways involved in CD4+TRM establishment have not been defined. Here, we performed transcriptional profiling of influenza-specific lung CD4+TRM following influenza infection to identify pathways implicated in CD4+TRM generation and homeostasis. Lung CD4+TRM displayed a unique transcriptional profile distinct from spleen memory, including up-regulation of a gene network induced by the transcription factor IRF4, a known regulator of effector T cell differentiation. In addition, the gene expression profile of lung CD4+TRM was enriched in gene sets previously described in tissue-resident regulatory T cells. Up-regulation of immunomodulatory molecules such as CTLA-4, PD-1, and ICOS, suggested a potential regulatory role for CD4+TRM in tissues. Using loss-of-function genetic experiments in mice, we demonstrate that IRF4 is required for the generation of lung-localized pathogen-specific effector CD4+T cells during acute influenza infection. Influenza-specific IRF4−/− T cells failed to fully express CD44, and maintained high levels of CD62L compared to wild type, suggesting a defect in complete differentiation into lung-tropic effector T cells. -

Molecular Profiling of Peripheral Blood Is Associated with Circulating Tumor Cells Content and Poor Survival in Metastatic Castration-Resistant Prostate Cancer
www.impactjournals.com/oncotarget/ Oncotarget, Vol. 6, No. 12 Molecular profiling of peripheral blood is associated with circulating tumor cells content and poor survival in metastatic castration-resistant prostate cancer Mercedes Marín-Aguilera1, Òscar Reig1,2, Juan José Lozano3, Natalia Jiménez1, Susana García-Recio1,4, Nadina Erill5, Lydia Gaba2, Andrea Tagliapietra2, Vanesa Ortega2, Gemma Carrera6, Anna Colomer5, Pedro Gascón4 and Begoña Mellado1,2 1 Translational Genomics Group and Targeted Therapeutics in Solid Tumors Group, Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), Barcelona, Spain 2 Medical Oncology Department, Hospital Clínic, Barcelona, Spain 3 Bioinformatics Platform Department, Centro de Investigación Biomédica en Red en el Área temática de Enfermedades Hepáticas y Digestivas (CIBEREHD), Hospital Clínic, Barcelona, Spain 4 Laboratory of Translational Oncology, Fundació Clínic per a la Recerca Biomèdica, Barcelona, Spain 5 Althia, Barcelona, Spain 6 Medical Oncology Department, Hospital Plató, Barcelona, Spain Correspondence to: Begoña Mellado, email: [email protected] Keywords: circulating tumor cells, peripheral blood, microarrays, cell search system Received: January 22, 2015 Accepted: February 14, 2015 Published: March 12, 2015 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT The enumeration of circulating -
![Downloaded from [266]](https://docslib.b-cdn.net/cover/7352/downloaded-from-266-347352.webp)
Downloaded from [266]
Patterns of DNA methylation on the human X chromosome and use in analyzing X-chromosome inactivation by Allison Marie Cotton B.Sc., The University of Guelph, 2005 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in The Faculty of Graduate Studies (Medical Genetics) THE UNIVERSITY OF BRITISH COLUMBIA (Vancouver) January 2012 © Allison Marie Cotton, 2012 Abstract The process of X-chromosome inactivation achieves dosage compensation between mammalian males and females. In females one X chromosome is transcriptionally silenced through a variety of epigenetic modifications including DNA methylation. Most X-linked genes are subject to X-chromosome inactivation and only expressed from the active X chromosome. On the inactive X chromosome, the CpG island promoters of genes subject to X-chromosome inactivation are methylated in their promoter regions, while genes which escape from X- chromosome inactivation have unmethylated CpG island promoters on both the active and inactive X chromosomes. The first objective of this thesis was to determine if the DNA methylation of CpG island promoters could be used to accurately predict X chromosome inactivation status. The second objective was to use DNA methylation to predict X-chromosome inactivation status in a variety of tissues. A comparison of blood, muscle, kidney and neural tissues revealed tissue-specific X-chromosome inactivation, in which 12% of genes escaped from X-chromosome inactivation in some, but not all, tissues. X-linked DNA methylation analysis of placental tissues predicted four times higher escape from X-chromosome inactivation than in any other tissue. Despite the hypomethylation of repetitive elements on both the X chromosome and the autosomes, no changes were detected in the frequency or intensity of placental Cot-1 holes. -

Macropinocytosis Requires Gal-3 in a Subset of Patient-Derived Glioblastoma Stem Cells
ARTICLE https://doi.org/10.1038/s42003-021-02258-z OPEN Macropinocytosis requires Gal-3 in a subset of patient-derived glioblastoma stem cells Laetitia Seguin1,8, Soline Odouard2,8, Francesca Corlazzoli 2,8, Sarah Al Haddad2, Laurine Moindrot2, Marta Calvo Tardón3, Mayra Yebra4, Alexey Koval5, Eliana Marinari2, Viviane Bes3, Alexandre Guérin 6, Mathilde Allard2, Sten Ilmjärv6, Vladimir L. Katanaev 5, Paul R. Walker3, Karl-Heinz Krause6, Valérie Dutoit2, ✉ Jann N. Sarkaria 7, Pierre-Yves Dietrich2 & Érika Cosset 2 Recently, we involved the carbohydrate-binding protein Galectin-3 (Gal-3) as a druggable target for KRAS-mutant-addicted lung and pancreatic cancers. Here, using glioblastoma patient-derived stem cells (GSCs), we identify and characterize a subset of Gal-3high glio- 1234567890():,; blastoma (GBM) tumors mainly within the mesenchymal subtype that are addicted to Gal-3- mediated macropinocytosis. Using both genetic and pharmacologic inhibition of Gal-3, we showed a significant decrease of GSC macropinocytosis activity, cell survival and invasion, in vitro and in vivo. Mechanistically, we demonstrate that Gal-3 binds to RAB10, a member of the RAS superfamily of small GTPases, and β1 integrin, which are both required for macro- pinocytosis activity and cell survival. Finally, by defining a Gal-3/macropinocytosis molecular signature, we could predict sensitivity to this dependency pathway and provide proof-of- principle for innovative therapeutic strategies to exploit this Achilles’ heel for a significant and unique subset of GBM patients. 1 University Côte d’Azur, CNRS UMR7284, INSERM U1081, Institute for Research on Cancer and Aging (IRCAN), Nice, France. 2 Laboratory of Tumor Immunology, Department of Oncology, Center for Translational Research in Onco-Hematology, Swiss Cancer Center Léman (SCCL), Geneva University Hospitals, University of Geneva, Geneva, Switzerland. -

Downloads/ (Accessed on 17 January 2020)
cells Review Novel Approaches for Identifying the Molecular Background of Schizophrenia Arkadiy K. Golov 1,2,*, Nikolay V. Kondratyev 1 , George P. Kostyuk 3 and Vera E. Golimbet 1 1 Mental Health Research Center, 34 Kashirskoye shosse, 115522 Moscow, Russian; [email protected] (N.V.K.); [email protected] (V.E.G.) 2 Institute of Gene Biology, Russian Academy of Sciences, 34/5 Vavilova Street, 119334 Moscow, Russian 3 Alekseev Psychiatric Clinical Hospital No. 1, 2 Zagorodnoye shosse, 115191 Moscow, Russian; [email protected] * Correspondence: [email protected] Received: 5 November 2019; Accepted: 16 January 2020; Published: 18 January 2020 Abstract: Recent advances in psychiatric genetics have led to the discovery of dozens of genomic loci associated with schizophrenia. However, a gap exists between the detection of genetic associations and understanding the underlying molecular mechanisms. This review describes the basic approaches used in the so-called post-GWAS studies to generate biological interpretation of the existing population genetic data, including both molecular (creation and analysis of knockout animals, exploration of the transcriptional effects of common variants in human brain cells) and computational (fine-mapping of causal variability, gene set enrichment analysis, partitioned heritability analysis) methods. The results of the crucial studies, in which these approaches were used to uncover the molecular and neurobiological basis of the disease, are also reported. Keywords: schizophrenia; GWAS; causal genetic variants; enhancers; brain epigenomics; genome/epigenome editing 1. Introduction Schizophrenia is a severe mental illness that affects between 0.5% and 0.7% of the human population [1]. Both environmental and genetic factors are thought to be involved in its pathogenesis, with genetic factors playing a key role in disease risk, as the heritability of schizophrenia is estimated to be 70–85% [2,3]. -

Identification of Potential Key Genes and Pathway Linked with Sporadic Creutzfeldt-Jakob Disease Based on Integrated Bioinformatics Analyses
medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Identification of potential key genes and pathway linked with sporadic Creutzfeldt-Jakob disease based on integrated bioinformatics analyses Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Abstract Sporadic Creutzfeldt-Jakob disease (sCJD) is neurodegenerative disease also called prion disease linked with poor prognosis. The aim of the current study was to illuminate the underlying molecular mechanisms of sCJD. The mRNA microarray dataset GSE124571 was downloaded from the Gene Expression Omnibus database. Differentially expressed genes (DEGs) were screened. -

Network-Based Prediction of Polygenic Disease Genes Involved in Cell Motility Miriam Bern†, Alexander King†, Derek A
Bern et al. BMC Bioinformatics 2019, 20(Suppl 12):313 https://doi.org/10.1186/s12859-019-2834-1 RESEARCH Open Access Network-based prediction of polygenic disease genes involved in cell motility Miriam Bern†, Alexander King†, Derek A. Applewhite and Anna Ritz* From International Workshop on Computational Network Biology: Modeling, Analysis and Control Washington, D.C., USA. 29 August 2018 Abstract Background: Schizophrenia and autism are examples of polygenic diseases caused by a multitude of genetic variants, many of which are still poorly understood. Recently, both diseases have been associated with disrupted neuron motility and migration patterns, suggesting that aberrant cell motility is a phenotype for these neurological diseases. Results: We formulate the POLYGENIC DISEASE PHENOTYPE Problem which seeks to identify candidate disease genes that may be associated with a phenotype such as cell motility. We present a machine learning approach to solve this problem for schizophrenia and autism genes within a brain-specific functional interaction network. Our method outperforms peer semi-supervised learning approaches, achieving better cross-validation accuracy across different sets of gold-standard positives. We identify top candidates for both schizophrenia and autism, and select six genes labeled as schizophrenia positives that are predicted to be associated with cell motility for follow-up experiments. Conclusions: Candidate genes predicted by our method suggest testable hypotheses about these genes’ role in cell motility regulation, offering a framework for generating predictions for experimental validation. Keywords: Semi-supervised learning, Functional interaction network, Schizophrenia, Autism, Cell motility Background participants [4, 5], indicating that schizophrenia arises Many polygenic diseases, which arise from the action from a multitude of cellular perturbations compounding or influence of multiple genes, are difficult to geneti- their effects. -

Viewed and Published Immediately Upon Acceptance Cited in Pubmed and Archived on Pubmed Central Yours — You Keep the Copyright
BMC Genomics BioMed Central Research article Open Access Differential gene expression in ADAM10 and mutant ADAM10 transgenic mice Claudia Prinzen1, Dietrich Trümbach2, Wolfgang Wurst2, Kristina Endres1, Rolf Postina1 and Falk Fahrenholz*1 Address: 1Johannes Gutenberg-University, Institute of Biochemistry, Mainz, Johann-Joachim-Becherweg 30, 55128 Mainz, Germany and 2Helmholtz Zentrum München – German Research Center for Environmental Health, Institute for Developmental Genetics, Ingolstädter Landstraße 1, 85764 Neuherberg, Germany Email: Claudia Prinzen - [email protected]; Dietrich Trümbach - [email protected]; Wolfgang Wurst - [email protected]; Kristina Endres - [email protected]; Rolf Postina - [email protected]; Falk Fahrenholz* - [email protected] * Corresponding author Published: 5 February 2009 Received: 19 June 2008 Accepted: 5 February 2009 BMC Genomics 2009, 10:66 doi:10.1186/1471-2164-10-66 This article is available from: http://www.biomedcentral.com/1471-2164/10/66 © 2009 Prinzen et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: In a transgenic mouse model of Alzheimer disease (AD), cleavage of the amyloid precursor protein (APP) by the α-secretase ADAM10 prevented amyloid plaque formation, and alleviated cognitive deficits. Furthermore, ADAM10 overexpression increased the cortical synaptogenesis. These results suggest that upregulation of ADAM10 in the brain has beneficial effects on AD pathology. Results: To assess the influence of ADAM10 on the gene expression profile in the brain, we performed a microarray analysis using RNA isolated from brains of five months old mice overexpressing either the α-secretase ADAM10, or a dominant-negative mutant (dn) of this enzyme. -

In This Table Protein Name, Uniprot Code, Gene Name P-Value
Supplementary Table S1: In this table protein name, uniprot code, gene name p-value and Fold change (FC) for each comparison are shown, for 299 of the 301 significantly regulated proteins found in both comparisons (p-value<0.01, fold change (FC) >+/-0.37) ALS versus control and FTLD-U versus control. Two uncharacterized proteins have been excluded from this list Protein name Uniprot Gene name p value FC FTLD-U p value FC ALS FTLD-U ALS Cytochrome b-c1 complex P14927 UQCRB 1.534E-03 -1.591E+00 6.005E-04 -1.639E+00 subunit 7 NADH dehydrogenase O95182 NDUFA7 4.127E-04 -9.471E-01 3.467E-05 -1.643E+00 [ubiquinone] 1 alpha subcomplex subunit 7 NADH dehydrogenase O43678 NDUFA2 3.230E-04 -9.145E-01 2.113E-04 -1.450E+00 [ubiquinone] 1 alpha subcomplex subunit 2 NADH dehydrogenase O43920 NDUFS5 1.769E-04 -8.829E-01 3.235E-05 -1.007E+00 [ubiquinone] iron-sulfur protein 5 ARF GTPase-activating A0A0C4DGN6 GIT1 1.306E-03 -8.810E-01 1.115E-03 -7.228E-01 protein GIT1 Methylglutaconyl-CoA Q13825 AUH 6.097E-04 -7.666E-01 5.619E-06 -1.178E+00 hydratase, mitochondrial ADP/ATP translocase 1 P12235 SLC25A4 6.068E-03 -6.095E-01 3.595E-04 -1.011E+00 MIC J3QTA6 CHCHD6 1.090E-04 -5.913E-01 2.124E-03 -5.948E-01 MIC J3QTA6 CHCHD6 1.090E-04 -5.913E-01 2.124E-03 -5.948E-01 Protein kinase C and casein Q9BY11 PACSIN1 3.837E-03 -5.863E-01 3.680E-06 -1.824E+00 kinase substrate in neurons protein 1 Tubulin polymerization- O94811 TPPP 6.466E-03 -5.755E-01 6.943E-06 -1.169E+00 promoting protein MIC C9JRZ6 CHCHD3 2.912E-02 -6.187E-01 2.195E-03 -9.781E-01 Mitochondrial 2- -
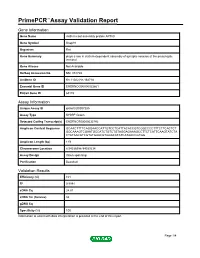
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name clathrin coat assembly protein AP180 Gene Symbol Snap91 Organism Rat Gene Summary plays a role in clathrin-dependent assembly of synaptic vesicles at the presynaptic terminal Gene Aliases Not Available RefSeq Accession No. NM_031728 UniGene ID Rn.11022,Rn.164718 Ensembl Gene ID ENSRNOG00000023861 Entrez Gene ID 65178 Assay Information Unique Assay ID qRnoCID0007285 Assay Type SYBR® Green Detected Coding Transcript(s) ENSRNOT00000032792 Amplicon Context Sequence GCAACTTTTCAGGAACCATTGTCCTCATTACACCGTCGGCCCCTTTCTTCACTCT GGCAAAGTCGAATGCCATCTGTCTGTAGGAGAAAGCCTTCTCATTCAAGTATCTA CTATAACGTCGTATGAACGTGGACATATCATAACCGTGG Amplicon Length (bp) 119 Chromosome Location 8:94036996-94039234 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 101 R2 0.9991 cDNA Cq 24.81 cDNA Tm (Celsius) 82 gDNA Cq Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report Snap91, Rat Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. Detected Coding Transcript(s) This is a list of the Ensembl transcript ID(s) that this assay will detect. -

Effects of Chronic Stress on Prefrontal Cortex Transcriptome in Mice Displaying Different Genetic Backgrounds
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Springer - Publisher Connector J Mol Neurosci (2013) 50:33–57 DOI 10.1007/s12031-012-9850-1 Effects of Chronic Stress on Prefrontal Cortex Transcriptome in Mice Displaying Different Genetic Backgrounds Pawel Lisowski & Marek Wieczorek & Joanna Goscik & Grzegorz R. Juszczak & Adrian M. Stankiewicz & Lech Zwierzchowski & Artur H. Swiergiel Received: 14 May 2012 /Accepted: 25 June 2012 /Published online: 27 July 2012 # The Author(s) 2012. This article is published with open access at Springerlink.com Abstract There is increasing evidence that depression signaling pathway (Clic6, Drd1a,andPpp1r1b). LA derives from the impact of environmental pressure on transcriptome affected by CMS was associated with genetically susceptible individuals. We analyzed the genes involved in behavioral response to stimulus effects of chronic mild stress (CMS) on prefrontal cor- (Fcer1g, Rasd2, S100a8, S100a9, Crhr1, Grm5,and tex transcriptome of two strains of mice bred for high Prkcc), immune effector processes (Fcer1g, Mpo,and (HA)and low (LA) swim stress-induced analgesia that Igh-VJ558), diacylglycerol binding (Rasgrp1, Dgke, differ in basal transcriptomic profiles and depression- Dgkg,andPrkcc), and long-term depression (Crhr1, like behaviors. We found that CMS affected 96 and 92 Grm5,andPrkcc) and/or coding elements of dendrites genes in HA and LA mice, respectively. Among genes (Crmp1, Cntnap4,andPrkcc) and myelin proteins with the same expression pattern in both strains after (Gpm6a, Mal,andMog). The results indicate significant CMS, we observed robust upregulation of Ttr gene contribution of genetic background to differences in coding transthyretin involved in amyloidosis, seizures, stress response gene expression in the mouse prefrontal stroke-like episodes, or dementia. -

Nº Ref Uniprot Proteína Péptidos Identificados Por MS/MS 1 P01024
Document downloaded from http://www.elsevier.es, day 26/09/2021. This copy is for personal use. Any transmission of this document by any media or format is strictly prohibited. Nº Ref Uniprot Proteína Péptidos identificados 1 P01024 CO3_HUMAN Complement C3 OS=Homo sapiens GN=C3 PE=1 SV=2 por 162MS/MS 2 P02751 FINC_HUMAN Fibronectin OS=Homo sapiens GN=FN1 PE=1 SV=4 131 3 P01023 A2MG_HUMAN Alpha-2-macroglobulin OS=Homo sapiens GN=A2M PE=1 SV=3 128 4 P0C0L4 CO4A_HUMAN Complement C4-A OS=Homo sapiens GN=C4A PE=1 SV=1 95 5 P04275 VWF_HUMAN von Willebrand factor OS=Homo sapiens GN=VWF PE=1 SV=4 81 6 P02675 FIBB_HUMAN Fibrinogen beta chain OS=Homo sapiens GN=FGB PE=1 SV=2 78 7 P01031 CO5_HUMAN Complement C5 OS=Homo sapiens GN=C5 PE=1 SV=4 66 8 P02768 ALBU_HUMAN Serum albumin OS=Homo sapiens GN=ALB PE=1 SV=2 66 9 P00450 CERU_HUMAN Ceruloplasmin OS=Homo sapiens GN=CP PE=1 SV=1 64 10 P02671 FIBA_HUMAN Fibrinogen alpha chain OS=Homo sapiens GN=FGA PE=1 SV=2 58 11 P08603 CFAH_HUMAN Complement factor H OS=Homo sapiens GN=CFH PE=1 SV=4 56 12 P02787 TRFE_HUMAN Serotransferrin OS=Homo sapiens GN=TF PE=1 SV=3 54 13 P00747 PLMN_HUMAN Plasminogen OS=Homo sapiens GN=PLG PE=1 SV=2 48 14 P02679 FIBG_HUMAN Fibrinogen gamma chain OS=Homo sapiens GN=FGG PE=1 SV=3 47 15 P01871 IGHM_HUMAN Ig mu chain C region OS=Homo sapiens GN=IGHM PE=1 SV=3 41 16 P04003 C4BPA_HUMAN C4b-binding protein alpha chain OS=Homo sapiens GN=C4BPA PE=1 SV=2 37 17 Q9Y6R7 FCGBP_HUMAN IgGFc-binding protein OS=Homo sapiens GN=FCGBP PE=1 SV=3 30 18 O43866 CD5L_HUMAN CD5 antigen-like OS=Homo