Quantum Effects in Radical B12 Enzymes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Anaerobic Radical Enzymes for Biotechnology
ChemBioEng Reviews Anaerobic radical enzymes for biotechnology Journal: ChemBioEng Reviews Manuscript ID cben.201800003.R1 Wiley - Manuscript type:For Review Peer Review Date Submitted by the Author: n/a Complete List of Authors: Jäger, Christof; University of Nottingham, Chemical and Environmental Engineering Croft, Anna; University of Nottingham, Chemical and Environmental Engineering Keywords: Radicals, Enzymes, Catalysis, Biotechnology, Anaerobic reactions Wiley-VCH Page 1 of 61 ChemBioEng Reviews 1 2 3 Christof M. Jäger* and Anna K. Croft* 4 5 6 7 8 9 Anaerobic radical enzymes for biotechnology 10 11 12 13 AUTHORS: Dr Christof Martin Jäger* and Dr Anna Kristina Croft* 14 15 16 ADDRESS: Department of Chemical and Environmental Engineering, University of 17 18 Nottingham, Nottingham, NG7 2RD, United Kingdom. [email protected], 19 For Peer Review 20 21 [email protected] 22 23 24 ABSTRACT: 25 26 27 28 Enzymes that proceed through radical intermediates have a rich chemistry that includes 29 30 functionalisation of otherwise unreactive carbon atoms, carbon-skeleton rearrangements, 31 32 aromatic reductions, and unusual eliminations. Especially under anaerobic conditions, 33 34 organisms have developed a wide range of approaches for managing these transformations 35 36 that can be exploited to generate new biological routes towards both bulk and specialty 37 38 39 chemicals. These routes are often either much more direct or allow access to molecules that 40 41 are inaccessible through standard (bio)chemical approaches. This review gives an overview 42 43 of some of the key enzymes in this area: benzoyl-CoA reductases (that effect the enzymatic 44 45 Birch reduction), ketyl radical dehydratases, coenzyme B12-dependant enzymes, glycyl 46 47 radical enzymes, and radical SAM (AdoMet radical) enzymes. -

Computational Genome and Pathway Analysis of Halophilic Archaea
Computational Genome and Pathway Analysis of Halophilic Archaea Dissertation Michaela Falb aus Heiligenstadt (Eichsfeld) 2005 Dissertation zur Erlangung des Doktorgrades der Fakultät für Chemie und Pharmazie der Ludwig-Maximilians-Universität München Computational Genome and Pathway Analysis of Halophilic Archaea Michaela Falb aus Heiligenstadt (Eichsfeld) 2005 Erklärung Diese Dissertation wurde im Sinne von §13 Abs. 3 bzw. 4 der Promotionsordnung vom 29. Januar 1998 von Prof. Dr. Dieter Oesterhelt betreut. Ehrenwörtliche Versicherung Diese Dissertation wurde selbständig, ohne unerlaubte Hilfe erarbeitet. München, den 31. August 2005 Michaela Falb Dissertation eingereicht am: 01.09.2005 1. Gutachter: Prof. Dr. Dieter Oesterhelt 2. Gutachter: Prof. Dr. Erich Bornberg-Bauer Mündliche Prüfung am: 22.12.2005 CONTENTS Summary 1 1 Introduction to Halophilic Archaea 3 1.1 Hypersaline environments 3 1.2 Taxononomy of halophilic archaea 5 1.3 Information processing in archaea 7 1.4 Physiology and metabolism of halophilic archaea 9 1.4.1 Osmotic adaptation 9 1.4.2 Nutritional demands, nutrient transport and sensing 10 1.4.3 Energy metabolism 11 1.5 Genomes of halophilic archaea 13 1.6 Motivation 15 2 Gene Prediction and Start Codon Selection in Halophilic Genomes 17 2.1 Introduction 17 2.2 Post-processing of gene prediction results by expert validation 19 2.3 Intrinsic features of haloarchaeal proteins and gene context analysis 22 2.3.1 Isoelectric points and amino acid distribution of halophilic proteins 22 2.3.2 Development of a pI scanning tool -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S. -

POLSKIE TOWARZYSTWO BIOCHEMICZNE Postępy Biochemii
POLSKIE TOWARZYSTWO BIOCHEMICZNE Postępy Biochemii http://rcin.org.pl WSKAZÓWKI DLA AUTORÓW Kwartalnik „Postępy Biochemii” publikuje artykuły monograficzne omawiające wąskie tematy, oraz artykuły przeglądowe referujące szersze zagadnienia z biochemii i nauk pokrewnych. Artykuły pierwszego typu winny w sposób syntetyczny omawiać wybrany temat na podstawie możliwie pełnego piśmiennictwa z kilku ostatnich lat, a artykuły drugiego typu na podstawie piśmiennictwa z ostatnich dwu lat. Objętość takich artykułów nie powinna przekraczać 25 stron maszynopisu (nie licząc ilustracji i piśmiennictwa). Kwartalnik publikuje także artykuły typu minireviews, do 10 stron maszynopisu, z dziedziny zainteresowań autora, opracowane na podstawie najnow szego piśmiennictwa, wystarczającego dla zilustrowania problemu. Ponadto kwartalnik publikuje krótkie noty, do 5 stron maszynopisu, informujące o nowych, interesujących osiągnięciach biochemii i nauk pokrewnych, oraz noty przybliżające historię badań w zakresie różnych dziedzin biochemii. Przekazanie artykułu do Redakcji jest równoznaczne z oświadczeniem, że nadesłana praca nie była i nie będzie publikowana w innym czasopiśmie, jeżeli zostanie ogłoszona w „Postępach Biochemii”. Autorzy artykułu odpowiadają za prawidłowość i ścisłość podanych informacji. Autorów obowiązuje korekta autorska. Koszty zmian tekstu w korekcie (poza poprawieniem błędów drukarskich) ponoszą autorzy. Artykuły honoruje się według obowiązujących stawek. Autorzy otrzymują bezpłatnie 25 odbitek swego artykułu; zamówienia na dodatkowe odbitki (płatne) należy zgłosić pisemnie odsyłając pracę po korekcie autorskiej. Redakcja prosi autorów o przestrzeganie następujących wskazówek: Forma maszynopisu: maszynopis pracy i wszelkie załączniki należy nadsyłać w dwu egzem plarzach. Maszynopis powinien być napisany jednostronnie, z podwójną interlinią, z marginesem ok. 4 cm po lewej i ok. 1 cm po prawej stronie; nie może zawierać więcej niż 60 znaków w jednym wierszu nie więcej niż 30 wierszy na stronie zgodnie z Normą Polską. -

All Enzymes in BRENDA™ the Comprehensive Enzyme Information System
All enzymes in BRENDA™ The Comprehensive Enzyme Information System http://www.brenda-enzymes.org/index.php4?page=information/all_enzymes.php4 1.1.1.1 alcohol dehydrogenase 1.1.1.B1 D-arabitol-phosphate dehydrogenase 1.1.1.2 alcohol dehydrogenase (NADP+) 1.1.1.B3 (S)-specific secondary alcohol dehydrogenase 1.1.1.3 homoserine dehydrogenase 1.1.1.B4 (R)-specific secondary alcohol dehydrogenase 1.1.1.4 (R,R)-butanediol dehydrogenase 1.1.1.5 acetoin dehydrogenase 1.1.1.B5 NADP-retinol dehydrogenase 1.1.1.6 glycerol dehydrogenase 1.1.1.7 propanediol-phosphate dehydrogenase 1.1.1.8 glycerol-3-phosphate dehydrogenase (NAD+) 1.1.1.9 D-xylulose reductase 1.1.1.10 L-xylulose reductase 1.1.1.11 D-arabinitol 4-dehydrogenase 1.1.1.12 L-arabinitol 4-dehydrogenase 1.1.1.13 L-arabinitol 2-dehydrogenase 1.1.1.14 L-iditol 2-dehydrogenase 1.1.1.15 D-iditol 2-dehydrogenase 1.1.1.16 galactitol 2-dehydrogenase 1.1.1.17 mannitol-1-phosphate 5-dehydrogenase 1.1.1.18 inositol 2-dehydrogenase 1.1.1.19 glucuronate reductase 1.1.1.20 glucuronolactone reductase 1.1.1.21 aldehyde reductase 1.1.1.22 UDP-glucose 6-dehydrogenase 1.1.1.23 histidinol dehydrogenase 1.1.1.24 quinate dehydrogenase 1.1.1.25 shikimate dehydrogenase 1.1.1.26 glyoxylate reductase 1.1.1.27 L-lactate dehydrogenase 1.1.1.28 D-lactate dehydrogenase 1.1.1.29 glycerate dehydrogenase 1.1.1.30 3-hydroxybutyrate dehydrogenase 1.1.1.31 3-hydroxyisobutyrate dehydrogenase 1.1.1.32 mevaldate reductase 1.1.1.33 mevaldate reductase (NADPH) 1.1.1.34 hydroxymethylglutaryl-CoA reductase (NADPH) 1.1.1.35 3-hydroxyacyl-CoA -

(12) Patent Application Publication (10) Pub. No.: US 2015/0240226A1 Mathur Et Al
US 20150240226A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2015/0240226A1 Mathur et al. (43) Pub. Date: Aug. 27, 2015 (54) NUCLEICACIDS AND PROTEINS AND CI2N 9/16 (2006.01) METHODS FOR MAKING AND USING THEMI CI2N 9/02 (2006.01) CI2N 9/78 (2006.01) (71) Applicant: BP Corporation North America Inc., CI2N 9/12 (2006.01) Naperville, IL (US) CI2N 9/24 (2006.01) CI2O 1/02 (2006.01) (72) Inventors: Eric J. Mathur, San Diego, CA (US); CI2N 9/42 (2006.01) Cathy Chang, San Marcos, CA (US) (52) U.S. Cl. CPC. CI2N 9/88 (2013.01); C12O 1/02 (2013.01); (21) Appl. No.: 14/630,006 CI2O I/04 (2013.01): CI2N 9/80 (2013.01); CI2N 9/241.1 (2013.01); C12N 9/0065 (22) Filed: Feb. 24, 2015 (2013.01); C12N 9/2437 (2013.01); C12N 9/14 Related U.S. Application Data (2013.01); C12N 9/16 (2013.01); C12N 9/0061 (2013.01); C12N 9/78 (2013.01); C12N 9/0071 (62) Division of application No. 13/400,365, filed on Feb. (2013.01); C12N 9/1241 (2013.01): CI2N 20, 2012, now Pat. No. 8,962,800, which is a division 9/2482 (2013.01); C07K 2/00 (2013.01); C12Y of application No. 1 1/817,403, filed on May 7, 2008, 305/01004 (2013.01); C12Y 1 1 1/01016 now Pat. No. 8,119,385, filed as application No. PCT/ (2013.01); C12Y302/01004 (2013.01); C12Y US2006/007642 on Mar. 3, 2006. -

Springer Handbook of Enzymes
Dietmar Schomburg Ida Schomburg (Eds.) Springer Handbook of Enzymes Alphabetical Name Index 1 23 © Springer-Verlag Berlin Heidelberg New York 2010 This work is subject to copyright. All rights reserved, whether in whole or part of the material con- cerned, specifically the right of translation, printing and reprinting, reproduction and storage in data- bases. The publisher cannot assume any legal responsibility for given data. Commercial distribution is only permitted with the publishers written consent. Springer Handbook of Enzymes, Vols. 1–39 + Supplements 1–7, Name Index 2.4.1.60 abequosyltransferase, Vol. 31, p. 468 2.7.1.157 N-acetylgalactosamine kinase, Vol. S2, p. 268 4.2.3.18 abietadiene synthase, Vol. S7,p.276 3.1.6.12 N-acetylgalactosamine-4-sulfatase, Vol. 11, p. 300 1.14.13.93 (+)-abscisic acid 8’-hydroxylase, Vol. S1, p. 602 3.1.6.4 N-acetylgalactosamine-6-sulfatase, Vol. 11, p. 267 1.2.3.14 abscisic-aldehyde oxidase, Vol. S1, p. 176 3.2.1.49 a-N-acetylgalactosaminidase, Vol. 13,p.10 1.2.1.10 acetaldehyde dehydrogenase (acetylating), Vol. 20, 3.2.1.53 b-N-acetylgalactosaminidase, Vol. 13,p.91 p. 115 2.4.99.3 a-N-acetylgalactosaminide a-2,6-sialyltransferase, 3.5.1.63 4-acetamidobutyrate deacetylase, Vol. 14,p.528 Vol. 33,p.335 3.5.1.51 4-acetamidobutyryl-CoA deacetylase, Vol. 14, 2.4.1.147 acetylgalactosaminyl-O-glycosyl-glycoprotein b- p. 482 1,3-N-acetylglucosaminyltransferase, Vol. 32, 3.5.1.29 2-(acetamidomethylene)succinate hydrolase, p. 287 Vol. -

Major Discussions and Tabular Citations Are Noted in Bold Type
Index Major discussions and tabular citations are noted in bold type. A acetyl enzyme 75-76 exchange reactions 75 Aasa, R. 41,42,58 kinetics 75 Abbott, S.l. 97 mechanism 75-76 Abdullah, M. 111 Acetyl-CoA carboxylase 220 Abeles, R.H. 9,24,28,34,54,55, 79, Acetyl-CoA synthetase 208-209,220 80, 151, 177, 178, 181, 185, 189, acetyl adenylate 208-209 190, 201 acetyl enzyme 208-209 Abramovitz, A.S. 77 exchange reaction 208 - 209 Abramson, R. 112 inversion of configuration on Acetaldehyde 177 -179 phosphorus 209 hydration 172 mechanism 208 - 209 Acetate Co A-transferase 108 triple-displacement reaction 209 Acetate kinase 92-96, 108 Acetylesterase 147 exchange reactions 93 N-Acetylglucosamine 89-90 kinetics 94 Acetylglucosamine phosphomutase mechanism 95 108 mercuric ion, effects on 94, 96 f3-N- Acetylglucosaminidase 147 noncontiguous binding of Acetylium ion 18 substrates 94-96 N-Acetylneuraminate lyase 182 nuc1eosidediphosphate kinase Acetyl phosphate 92-96, 202, 217 activity 93-94 Acid phosphatase 120-121, 147 phosphoenzyme 92-96 phosphoenzyme 120 steric inversion on phosphorus 92 Acid protease from Rhizopus surface walk 96 chinensis 149 triple displacement 92, 95 Aconitase 182 Acetoacetate decarboxylase 17, Aconitate ~-isomerase 200 157 -158, 182 Acrosin 148 exchange reactions 157 -158 Actinidin 148 mechanism 157 Activation of water 114, 119, 130-131, Schiff base intermediates 157 145-146,173,177 Acetoacetate, decarboxylation, [Acyl-carrier protein] nonenzymic 17 acetyltransferase 106 Acetoacetyl-CoA 75, 104-106, [Acy I-carrier -protein] 166-167 malonyltransferase 76-77, 106 Acetolactate synthase 182 malonyl enzyme 76-77 Acetyl adenylate 203 Acyl-CoA dehydrogenase 49 Acetylcholinesterase 147 Acyl-CoA hydrolase 147 Acetyl-CoA 17,74,75,76, 166, 167, Adachi, K., 56 168-170, 208, 209, 214 Adair, Jr., W.L. -
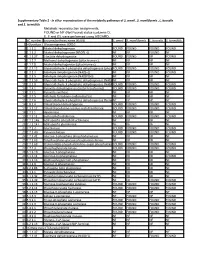
Supplementary Table 2 - in Silico Reconstruction of the Metabolic Pathways of S
Supplementary Table 2 - In silico reconstruction of the metabolic pathways of S. amnii , S. moniliformis , L. buccalis and S. termiditis Metabolic reconstruction assignments, FOUND or NF (Not Found) status (columns D, E, F and G), were performed using ASGARD, EC number Enzyme/pathway name (KEGG) S. amnii S. moniliformis L. buccalis S. termiditis 1 >Glycolysis / Gluconeogenesis 00010 2 1.1.1.1 Alcohol dehydrogenase. FOUND FOUND FOUND FOUND 3 1.1.1.2 Alcohol dehydrogenase (NADP(+)). NF NF FOUND NF 4 1.1.1.27 L-lactate dehydrogenase. FOUND FOUND NF FOUND 5 1.1.2.7 Methanol dehydrogenase (cytochrome c). NF NF NF NF 6 1.1.2.8 Alcohol dehydrogenase (cytochrome c). NF NF NF NF 7 1.2.1.12 Glyceraldehyde-3-phosphate dehydrogenase (phosphorylating).FOUND FOUND FOUND FOUND 8 1.2.1.3 Aldehyde dehydrogenase (NAD(+)). NF NF FOUND FOUND 9 1.2.1.5 Aldehyde dehydrogenase (NAD(P)(+)). NF NF NF NF 10 1.2.1.59 Glyceraldehyde-3-phosphate dehydrogenase (NAD(P)(+))NF (phosphorylating).NF NF NF 11 1.2.1.9 Glyceraldehyde-3-phosphate dehydrogenase (NADP(+)).FOUND FOUND FOUND FOUND 12 1.2.4.1 Pyruvate dehydrogenase (acetyl-transferring). FOUND FOUND FOUND FOUND 13 1.2.7.1 Pyruvate synthase. NF NF NF NF 14 1.2.7.5 Aldehyde ferredoxin oxidoreductase. NF NF NF NF 15 1.2.7.6 Glyceraldehyde-3-phosphate dehydrogenase (ferredoxin).NF NF NF NF 16 1.8.1.4 Dihydrolipoyl dehydrogenase. FOUND FOUND FOUND FOUND 17 2.3.1.12 Dihydrolipoyllysine-residue acetyltransferase. FOUND FOUND FOUND FOUND 18 2.7.1.1 Hexokinase. -

Searching for Metabolic Pathways of Anaerobic Digestion: a Useful List
Chapter Searching for Metabolic Pathways of Anaerobic Digestion: A Useful List of the Key Enzymes Anna Sikora, Anna Detman, Damian Mielecki, Aleksandra Chojnacka and Mieczysław Błaszczyk Abstract The general scheme of anaerobic digestion is well known. It is a complex process promoted by the interaction of many groups of microorganisms and has four major steps: hydrolysis, acidogenesis, acetogenesis, and methanogenesis. The aim of the study was to prepare a systematized list of the selected enzymes responsible for the key pathways of anaerobic digestion based on the Kyoto Encyclopedia of Genes and Genomes database resource. The list contains (i) key groups of hydrolases involved in the process of degradation of organic matter; (ii) the enzymes catalyzing reac- tions leading to pyruvate formation; (iii) the enzymes of metabolic pathways of further pyruvate transformations; (iv) the enzymes of glycerol transformations; (v) the enzymes involved in transformation of gaseous or nongaseous products of acidic fermentations resulting from nonsyntrophic nutritional interactions between microbes; (vi) the enzymes of amino acid fermentations; (vii) the enzymes involved in acetogenesis; and (viii) the enzymes of the recognized pathways of methanogenesis. Searching for the presence and activity of the enzymes as well as linking structure and function of microbial communities allows to develop a funda- mental understanding of the processes, leading to methane production. In this contribution, the present study is believed to be a piece to the enzymatic road map of anaerobic digestion research. Keywords: anaerobic digestion, enzymes, hydrolysis, acidogenesis, acetogenesis, methanogenesis, syntrophy, metabolic pathways 1. Introduction Anaerobic digestion (AD), whose final products are methane and carbon diox- ide, is a common process in natural anoxic environments such as water sediments, wetlands, or marshlands. -

The Enzyme List Class 5 — Isomerases
The Enzyme List Class 5 — Isomerases Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB) LATEX version prepared by Andrew McDonald, School of Biochemistry and Immunology, Trinity College Dublin, Ireland Generated from the ExplorEnz database, March 2019 © 2019 IUBMB Contents EC 5.1 Racemases and epimerases 2 EC 5.1.1 Acting on amino acids and derivatives......................................2 EC 5.1.2 Acting on hydroxy acids and derivatives.....................................6 EC 5.1.3 Acting on carbohydrates and derivatives.....................................8 EC 5.1.99 Acting on other compounds........................................... 17 EC 5.2 cis-trans-Isomerases 19 EC 5.2.1 cis-trans Isomerases (only sub-subclass identified to date)............................ 19 EC 5.3 Intramolecular oxidoreductases 21 EC 5.3.1 Interconverting aldoses and ketoses, and related compounds........................... 21 EC 5.3.2 Interconverting keto- and enol-groups...................................... 28 EC 5.3.3 Transposing C=C bonds.............................................. 30 EC 5.3.4 Transposing S-S bonds.............................................. 35 EC 5.3.99 Other intramolecular oxidoreductases...................................... 35 EC 5.4 Intramolecular transferases 38 EC 5.4.1 Transferring acyl groups............................................. 38 EC 5.4.2 Phosphotransferases (phosphomutases)...................................... 39 EC 5.4.3 Transferring amino groups........................................... -

Supplementary Table S1. Summary of MEROPS and ESTHER Database Description for Peptidases and Lipases, Respectively
Supplementary Table S1. Summary of MEROPS and ESTHER database description for peptidases and lipases, respectively. Peptidases MEROPS ID MEROPS description summary Selected for further analyses A8 production of the bacterial cell wall No M1 some are cleaving amino acids from small peptides Yes M3 Lactobacillus oligoendopeptidase cleaves oligopeptides Yes M14 digestion of food and other functions Yes M20 conversion of proteins to free amino acids. Yes M24 removes N-terminal Methionine from protein Yes M28 alkaline phosphatase isozyme conversion No M29 catabolic peptidase in Streptococcus thermophilus Yes M42 used by Lactobacillus for nutrition on milk-protein powder Yes M48 degradation of abnormal proteins No M50 regulated intermembrane proetolysis No S8 probably involved in nutrition Yes S12 synthesis and remodelling of bacterial cell walls No S15 degradation of casein Yes S24 regulatory stress response No S26 remove the signal peptides and facilitate secretion No Lipases ESTHER family ESTHER description summary Selected for further analyses 6_AlphaBeta_hydrolase not well characterized No Abhydrolase_7 acetyl-esterase_deacetylase or Dienelactone_hydrolase or xylan esterase No Bacterial_EstLip_FamX Family of Bacterial lipolytic enzymes Yes Carb_B_Bacteria carboxylesterase, type B Yes CarbLipBact_2 members of this group are esterases, lipases Yes Duf_1023 proteins of unknown function No Duf_3089 lipolytic enzyme family defined from isolation and characterization of two esterases from a metagenomic library Yes Duf_900 proteins of unknown function No Epoxide_hydrolase conversion of epoxides to corresponding diols No GTSAGmotif lipases from metagenomic library Yes Hormone-sensitive_lipase_like esterase/lipase, family IV Yes Lipase_2 lipases Yes Lipase_3 lipases Yes Monoglyceridelipase_lysophospholip has some homology to peptidases S33 No NFM-deformylase N-formylmaleamic acid to formic and maleamic acid No PGAP1 attachment to proteins factor 1 No Supplementary Table S2.