Morphologic Changes in Red Blood Cells: an Illustrated Review of Clinically Important Light Microscopic Findings
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
![Anormal Rbc in Peripheral Blood. [Repaired].Pdf](https://docslib.b-cdn.net/cover/4277/anormal-rbc-in-peripheral-blood-repaired-pdf-544277.webp)
Anormal Rbc in Peripheral Blood. [Repaired].Pdf
1. Acanthocyte 2. Burr-cell 3. Microcyte 1. Basophilic Normoblast 2. Polychromatic Normoblast 3. Pycnotic Normoblast 4. Plasmocyte 5. Eosinophil 6. Promyelocyte 1. Macrocyte 2. Elliptocyte 1. Microcyte 2. Normocyte 1. Polychromatic Erythrocyte 2. Acanthocyte 3. Elliptocyte 1. Polychromatic Normoblast 2. Pycnotic Normoblast 3. Neutrophil Myelocyte 4. Neutrophil Metamyelocyte 1. Schistocyte 2. Microcyte BASOPHILIC ( EARLY ) NORMOBLASTS Basophilic Erythroblast Basophilic Stippling, Blood smear, May-Giemsa stain, (×1000) CABOT'S RINGS Drepanocyte Elliptocyte Erythroblast ERYTHROBLAST in the blood Howell-jolly body Hypo chromic LACRYMOCYTES Leptocyte Malaria, Blood smear, May-Giemsa stain, ×1000 MICROCYTES Orthochromatic erythroblast Pappen heimer Bodies & 1. Schistocyte 2. Elliptocyte 3. Acanthocyte POIKILOCYTOSIS Polychromatic Erythroblast Pro Erytroblast Proerythroblasts Reticulocyte Rouleaux SICKLE CELLS Sickle cell Spherocyte Spherocyte Spherocyte SPHEROCYTES STOMATOCYTES Target Cells Tear Drop Cell, Blood smear, May-Giemsa stain, x1000 Anulocyte 1. Burr-cell 2. Elliptocyte 1. Macrocyte 2. Microcyte 3. Elliptocyte 4. Schistocyte 1. Ovalocyte 2. Lacrymocyte 3. Target cell 1. Polychromatic Erythrocyte 2. Basophilic Stippling 1. Proerythroblast 2. Basophilic Erythroblast 3. Intermediate Erythroblast 4. Late Erythroblast 5. Monocyte 6. Lymphocyte 1. Target-cell 2. Elliptocyte 3. Acanthocyte 4. Stomatocyte 5. Schistocyte 6. Polychromatophilic erythrocyte. 1.Pro erythroblast 2.Basophilic normoblast 3.Polychromatic normoblast 4.Pycnotic normoblast -

The Hematological Complications of Alcoholism
The Hematological Complications of Alcoholism HAROLD S. BALLARD, M.D. Alcohol has numerous adverse effects on the various types of blood cells and their functions. For example, heavy alcohol consumption can cause generalized suppression of blood cell production and the production of structurally abnormal blood cell precursors that cannot mature into functional cells. Alcoholics frequently have defective red blood cells that are destroyed prematurely, possibly resulting in anemia. Alcohol also interferes with the production and function of white blood cells, especially those that defend the body against invading bacteria. Consequently, alcoholics frequently suffer from bacterial infections. Finally, alcohol adversely affects the platelets and other components of the blood-clotting system. Heavy alcohol consumption thus may increase the drinker’s risk of suffering a stroke. KEY WORDS: adverse drug effect; AODE (alcohol and other drug effects); blood function; cell growth and differentiation; erythrocytes; leukocytes; platelets; plasma proteins; bone marrow; anemia; blood coagulation; thrombocytopenia; fibrinolysis; macrophage; monocyte; stroke; bacterial disease; literature review eople who abuse alcohol1 are at both direct and indirect. The direct in the number and function of WBC’s risk for numerous alcohol-related consequences of excessive alcohol increases the drinker’s risk of serious Pmedical complications, includ- consumption include toxic effects on infection, and impaired platelet produc- ing those affecting the blood (i.e., the the bone marrow; the blood cell pre- tion and function interfere with blood cursors; and the mature red blood blood cells as well as proteins present clotting, leading to symptoms ranging in the blood plasma) and the bone cells (RBC’s), white blood cells from a simple nosebleed to bleeding in marrow, where the blood cells are (WBC’s), and platelets. -
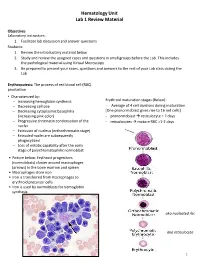
Hematology Unit Lab 1 Review Material
Hematology Unit Lab 1 Review Material Objectives Laboratory instructors: 1. Facilitate lab discussion and answer questions Students: 1. Review the introductory material below 2. Study and review the assigned cases and questions in small groups before the Lab. This includes the pathological material using Virtual Microscopy 3. Be prepared to present your cases, questions and answers to the rest of your Lab class during the Lab Erythropoiesis: The process of red blood cell (RBC) production • Characterized by: − Increasing hemoglobin synthesis Erythroid maturation stages (Below): − Decreasing cell size - Average of 4 cell divisions during maturation − Decreasing cytoplasmic basophilia [One pronormoblast gives rise to 16 red cells] (increasing pink color) - pronormoblast → reticulocyte = 7 days − Progressive chromatin condensation of the - reticulocytes → mature RBC =1-2 days nuclei − Extrusion of nucleus (orthochromatic stage) − Extruded nuclei are subsequently phagocytized − Loss of mitotic capability after the early stage of polychromatophilic normoblast • Picture below: Erythroid progenitors (normoblasts) cluster around macrophages (arrows) in the bone marrow and spleen • Macrophages store iron • Iron is transferred from macrophages to erythroid precursor cells • Iron is used by normoblasts for hemoglobin synthesis aka nucleated rbc aka reticulocyte 1 Mature Red Blood Cell 7-8 microns; round / ovoid biconcave disc with orange-red cytoplasm, no RNA, no nucleus; survives ~120 days in circulation Classification of Anemia by Morphology 1. -

Morphological Study of Human Blood for Different Diseases
Research Article ISSN: 2574 -1241 DOI: 10.26717/BJSTR.2020.30.004893 Morphological Study of Human Blood for Different Diseases Muzafar Shah1*, Haseena1, Kainat1, Noor Shaba1, Sania1, Sadia1, Akhtar Rasool2, Fazal Akbar2 and Muhammad Israr3 1Centre for Animal Sciences & Fisheries, University of Swat, Pakistan 2Centre for Biotechnology and Microbiology, University of Swat, Pakistan 3Department of Forensic Sciences, University of Swat, Pakistan *Corresponding author: Muzafar Shah, Centre for Animal Sciences & Fisheries, University of Swat, Pakistan ARTICLE INFO ABSTRACT Received: August 25, 2020 The aim of our study was the screening of blood cells on the basis of morphology for different diseased with Morphogenetic characters I e. ear lobe attachment, clinodactyly Published: September 07, 2020 and tongue rolling. For this purpose, 318 blood samples were collected randomly. Samples were examined under the compound microscopic by using 100x with standard Citation: Muzafar Shah, Haseena, method. The results show 63 samples were found normal while in 255 samples, different Kainat, Noor Shaba, Sania, Sadia, et al. types of morphological changes were observed which was 68.5%, in which Bite cell 36%, Morphological Study of Human Blood for Elliptocyte 34%, Tear drop cell 30%, Schistocyte 26%, Hypochromic cell 22.5%, Irregular Different Diseases. Biomed J Sci & Tech Res contracted cell 16%, Echinocytes 15.5%, Roleaux 8%, Boat shape 6.5%, Sickle cell 5%, Keratocyte 4% and Acanthocytes 1.5%. During the screening of slides, bite cell, elliptocyte, tear drop cell, schistocytes, hypochromic cell, irregular contracted cells were found 30(1)-2020.Keywords: BJSTR.Human MS.ID.004893. blood; Diseases; frequently while echinocytes, boat shape cell, acanthocytes, sickle cells and keratocytes Morphological; Acanthocytes; Keratocyte were found rarely. -

Complete Blood Count in Primary Care
Complete Blood Count in Primary Care bpac nz better medicine Editorial Team bpacnz Tony Fraser 10 George Street Professor Murray Tilyard PO Box 6032, Dunedin Clinical Advisory Group phone 03 477 5418 Dr Dave Colquhoun Michele Cray free fax 0800 bpac nz Dr Rosemary Ikram www.bpac.org.nz Dr Peter Jensen Dr Cam Kyle Dr Chris Leathart Dr Lynn McBain Associate Professor Jim Reid Dr David Reith Professor Murray Tilyard Programme Development Team Noni Allison Rachael Clarke Rebecca Didham Terry Ehau Peter Ellison Dr Malcolm Kendall-Smith Dr Anne Marie Tangney Dr Trevor Walker Dr Sharyn Willis Dave Woods Report Development Team Justine Broadley Todd Gillies Lana Johnson Web Gordon Smith Design Michael Crawford Management and Administration Kaye Baldwin Tony Fraser Kyla Letman Professor Murray Tilyard Distribution Zane Lindon Lyn Thomlinson Colleen Witchall All information is intended for use by competent health care professionals and should be utilised in conjunction with © May 2008 pertinent clinical data. Contents Key points/purpose 2 Introduction 2 Background ▪ Haematopoiesis - Cell development 3 ▪ Limitations of reference ranges for the CBC 4 ▪ Borderline abnormal results must be interpreted in clinical context 4 ▪ History and clinical examination 4 White Cells ▪ Neutrophils 5 ▪ Lymphocytes 9 ▪ Monocytes 11 ▪ Basophils 12 ▪ Eosinophils 12 ▪ Platelets 13 Haemoglobin and red cell indices ▪ Low haemoglobin 15 ▪ Microcytic anaemia 15 ▪ Normocytic anaemia 16 ▪ Macrocytic anaemia 17 ▪ High haemoglobin 17 ▪ Other red cell indices 18 Summary Table 19 Glossary 20 This resource is a consensus document, developed with haematology and general practice input. We would like to thank: Dr Liam Fernyhough, Haematologist, Canterbury Health Laboratories Dr Chris Leathart, GP, Christchurch Dr Edward Theakston, Haematologist, Diagnostic Medlab Ltd We would like to acknowledge their advice, expertise and valuable feedback on this document. -

Laboratory Diagnosis Review
Laboratory Diagnosis Review Hematology Definition: The study of the three cellular elements of blood: Red Blood Cells (RBCs), White Blood Cells (WBCs), and Platelets Hemoglobin (Hgb or Hb): The oxygen carrying compound in RBCs Reference Range: Men 14-18 g/dL, Women 12-16 g/dL, Boy and girl levels are equal till age 11 Smoking increases, Pregnancy decreases, Capillary levels in newborns are higher than venous levels, Race, Position, and Time of day have minor effects, High WBCs may falsely raise Hgb. Below normal Hgb = anemia Red Blood Cell Count Reference Range: Men 4l5-6 million / cubic ml, Women4.0-5.5 million / ml3. Hematocrit (Hct): The ratio of RBCs to plasma Reference Range, Men 40%-54%, Women 37%-47%, Depends mostly on the number of RBCs but is slightly effected by the average RBC size, Not measured directly, but is calculated from the RBC count and the mean corpuscular volume (MCV). Increased by smoking, Decrease = anemia Useful Relationships: Hb X 3 = Hct RBCs (millions) X 3 = Hgb RBCs (millions) X 9 = Hct Wintrobe Indices: These indices are only significant if the RBCs, Hgb, and/or Hct. is abnormal MCV: Mean Corpuscular Volume MCH: Mean Corpuscular Hemoglobin MCHC: Mean Corpuscular Hemoglobin Concentration RDW: Red blood cell Distribution Width MCV: Reference Ranges: Men 80-95 fl (femtoliters), Women 81-99 fl (femto = 1 quadrillionth) Increased MCV = Macrocytosis, Decreased = Microcytosis MCV is increased by smoking, by B12 and/or folic acid deficiency, chronic liver disease, chronic alcoholism, Cardiorespiratory problems… Some macrocytic patients will not have macrocytosis MCV is decreased by iron deficiency, thalassemia, and anemia of chronic disease MCH: Reference Range 27-31 pg MCH is increased by Macrocytic anemias. -

Identifying Peripheral Blood Leukocytes and Erythrocytes in a Patient with Iron Deficiency Anemia
ADVANCED BLOOD CELL ID: IDENTIFYING PERIPHERAL BLOOD LEUKOCYTES AND ERYTHROCYTES IN A PATIENT WITH IRON DEFICIENCY ANEMIA Educational commentary is provided for participants enrolled in program #259- Advanced Blood Cell Identification. This virtual blood cell identification program includes case studies with more difficult challenges. To view the blood cell images in more detail, click on the sample identification numbers underlined in the paragraphs below. This will open a virtual image of the selected cell and the surrounding fields. If the image opens in the same window as the commentary, saving the commentary PDF and opening it outside your browser will allow you to switch between the commentary and the images more easily. Click on this link for the API ImageViewerTM Instructions. Learning Outcomes After completion of this exercise, participants will be able to: • describe morphologic features of monocytes and lymphocytes, and • identify distinguishing morphologic features in red blood cells associated with iron deficiency anemia. Case Study A 78 year old female patient was seen by her primary care physician due to extreme fatigue and headaches. The CBC results are as follows: WBC=9.3 x 109/L, RBC=4.43 x 1012/L, Hgb=8.7 g/dL, Hct=26.1%, MCV=58.9 fL, MCH=19.6 pg, MCHC=33.3 g/dL, RDW=24.8%, Platelet=425 x 109/L. Educational Commentary The cells annotated for commentary in this advanced testing event were selected from the peripheral blood smear of an elderly woman diagnosed with iron deficiency anemia (IDA). IDA is a common worldwide disorder. It can be caused by lack of adequate dietary iron, the malabsorption of iron, increased need for iron as in pregnancy or infancy and, most often, by bleeding. -

10 11 Cyto Slides 81-85
NEW YORK STATE CYTOHEMATOLOGY PROFICIENCY TESTING PROGRAM Glass Slide Critique ~ November 2010 Slide 081 Diagnosis: MDS to AML 9 WBC 51.0 x 10 /L 12 Available data: RBC 3.39 x 10 /L 72 year-old female Hemoglobin 9.6 g/dL Hematocrit 29.1 % MCV 86.0 fL Platelet count 16 x 109 /L The significant finding in this case of Acute Myelogenous Leukemia (AML) was the presence of many blast forms. The participant median for blasts, all types was 88. The blast cells in this case (Image 081) are large, irregular in shape and contain large prominent nucleoli. It is difficult to identify a blast cell as a myeloblast without the presence of an Auer rod in the cytoplasm. Auer rods were reported by three participants. Two systems are used to classify AML into subtypes, the French- American-British (FAB) and the World Health Organization (WHO). Most are familiar with the FAB classification. The WHO classification system takes into consideration prognostic factors in classifying AML. These factors include cytogenetic test results, patient’s age, white blood cell count, pre-existing blood disorders and a history of treatment with chemotherapy and/or radiation therapy for a prior cancer. The platelet count in this case was 16,000. Reduced number of platelets was correctly reported by 346 (94%) of participants. Approximately eight percent of participants commented that the red blood cells in this case were difficult to evaluate due to the presence of a bluish hue around the red blood cells. Comments received included, “On slide 081 the morphology was difficult to evaluate since there was a large amount of protein surrounding RBC’s”, “Slide 081 unable to distinguish red cell morphology due to protein” and “Unable to adequately assess morphology on slide 081 due to poor stain”. -

Advanced Blood Cell Id: Peripheral Blood Findings in Sickle Cell Anemia
ADVANCED BLOOD CELL ID: PERIPHERAL BLOOD FINDINGS IN SICKLE CELL ANEMIA Educational commentary is provided for participants enrolled in program #259- Advanced Blood Cell Identification. This virtual blood cell identification program includes case studies with more difficult challenges. To view the blood cell images in more detail, click on the sample identification numbers underlined in the paragraphs below. This will open a virtual image of the selected cell and the surrounding fields. If the image opens in the same window as the commentary, saving the commentary PDF and opening it outside your browser will allow you to switch between the commentary and the images more easily. Click on this link for the API ImageViewerTM Instructions. Learning Outcomes After completing this exercise, participants should be able to: • describe morphologic features of normal peripheral blood leukocytes. • identify morphologic characteristics distinctive of sickle cells. • distinguish selected RBC inclusions based on morphologic features. • describe significant morphologic characteristics of nucleated red blood cells. Case Study The CBC from a 30 year old African American male is as follows: WBC=9.5 x 109/L, RBC=1.66 x 1012/L, Hgb=5.0 g/dL, Hct=13.9%, MCV=83.7 fL, MCH=30.1 pg, MCHC=36.0 g/dL, RDW-CV=24.9%, MPV=9.6 fL, Platelet=326 x 109/L. Educational Commentary The cells and RBC inclusions chosen for identification in this testing event were seen in the peripheral blood of a man with a severe anemia resulting from sickle cell disease. The cell shown in ABI-08 contains a Howell-Jolly body. -

Laboratory Interpretation: a Focus on WBC's, RBC's, and LFT's
Laboratory Interpretation: A Focus on WBC’s, RBC’s, and LFT’s Wendy L. Wright, MS, ANP-BC, FNP-BC, FAANP, FAAN, FNAP Adult/Family Nurse Practitioner Owner - Wright & Associates Family Healthcare Amherst, NH Owner – Wright & Associates Family Healthcare Concord, NH Owner – Partners in Healthcare Education, LLC Wright, 2019 1 1 Relevant Financial Relationship Disclosure Statement Title of talk • I will not discuss off label use and/or investigational use of any drugs/devices. • I don’t have the following relevant financial relationships to report in relationship to this presentation. Wright, 2019 2 2 Objectives • Upon completion of this lecture, the participant will be able to: – Identify a step approach to the interpretation of a cbc – rbc’s and wbc’s, and hepatic function tests – Discuss various laboratory abnormalities identified on an individual throughout the lifespan – Systematically interpret laboratory findings using case studies Wright, 2019 3 3 Wright, 2019 1 Red Blood Cell Formation • Formed in bone marrow (erythropoiesis) • When mature, the rbc is released into circulation • Mature rbc has a life span of approximately 120 days – Many factors trigger an increase in the production of rbc’s by the bone marrow, but a decrease in O2 is the most common. – Low tissue oxygen levels trigger the endothelial cells in the kidneys to secrete erythropoietin – which in turn, stimulates bone marrow red cell production Goodnough LT, Skikne B, Brugnara C. Erythropoietin, iron, and erythropoiesis. Blood. 2000;96:823-833. Wright, 2019 4 4 Anemia: -

Developing a Computer-Based Information System to Improve the Diagnosis of Blood Anemia
I I Developing a Computer-based Information System to Improve the Diagnosis of Blood Anemia By Bashar Abdallah Issa Khawaldeh Supervisor Dr. Basim Alhadidi This Thesis is submitted to the Department of Computer Information Systems, Faculty of Information Technology, Middle East University in partial fulfillment for the requirements for the degree of Master Degree in Computer Information System. Department of Computer Information Systems Faculty of Information Technology Middle East University (May 201 3) Amman – Jordan II III IV V VI ACKNOWLEDGMENTS I would like to thank my supervisor Dr. Basim Alhadidi for his support, encouragement, proofreading of thesis drafts, and helping me throughout my thesis, and so directing to the right track of Image processing. I thank the Information Technology Faculty members at the Middle East University for Graduate Studies; I thank my father and my mother for their continued support during my study. VII DEDICATION All praise belongs to Allah and all thanks to Allah. I dedicate this work to Parents, brothers, sisters, relatives, friends, and to all those who helped, supported and taught me. VIII Table of Contents Developing a Computer- based Information System to Improve the Diagnosis of Blood Anemia .…. I ………………………………….……..…................... .. ...... ………………...………………………..…….………. II Authorization Statement ………………………………………………….…………...………………………...…..…….……. III Examination Committee Decision ………………..…………………...…………………………………...……...…..…... IV Declaration ………………………………………………………………………………………………………………………….... -

Diagnosis from the Blood Smear
247 บทความฟื้นวิชา Diagnosis from the Blood Smear กิตติ ตอจรัส กองพยาธิวิทยา�รงพยาบา�พร�มงก��เก�า ปจจุบันมีการนำาเครื่องนับเม็ดเลือดอัตโนมัติ (automated สมมีการกระจายของเม็ดเลือดชนิดตางๆ อยางเหมาะสม ไมซอนกัน blood – cell analyzers) มาใชในการตรวจวิเคราะหทางหอง การตรวจสเมียรเลือด ผูตรวจตองดูดวยกลองใชกำาลังขยาย ปฏิบัติการซึ่งไดผลถูกตองและรวดเร็ว การตรวจสเมียรเลือด ต่ำา (10X10) กอนเสมอ เพื่อดูคุณภาพของสไลด (slide) การติด (blood smear) จึงถูกลดบทบาทลงไปนอยกวารอยละ 10 – 15 สีรวมทั้งจะไดเห็นภาพทั่วๆ ไป ของเม็ดเลือดแดง เม็ดเลือดขาว อยางไรก็ดี การตรวจสเมียรเลือดมีราคาถูกกวา แมวายังตองอาศัย และเกร็ดเลือด โดยจะทำาใหเห็นเม็ดเลือดขาวที่ผิดปกติไดงายขึ้น ขั้นตอนในการเตรียม ใชบุคลากรที่มีทักษะในการแปลผล แตยัง แลวจึงตรวจดูเม็ดเลือดทุกชนิดในสเมียรเลือดนั้น เปนเครื่องมือที่สำาคัญในการชวยการวินิจฉัยที่สำาคัญ (crucial รูปรางเม็ดเลือดแดง (red cell morphology) diagnosis aid)1 ตารางที่ 1 แสดงขอบงชี้ทางคลินิกที่ตองตรวจ เม็ดเลือดแดงปกติมีขนาด 7.2 – 7.9 µm. รูปรางเปน biconcave สเมียรเลือด เพื่อการวินิจฉัยเบื้องตน (provisional diagnosis) disc ขอบติดสี hemoglobin ตรงกลางไมติดสี (clear central เพื่อวินิจฉัยแยกโรค และการตรวจทางหองปฏิบัติการอื่นๆ ตอไป area) สามารถดูปริมาตร (MCV) ไดจากเครื่องนับเม็ดเลือดแดง หลักการวินิจฉัยจากการตรวจสเมียรเลือด อัตโนมัติ การดู blood smear จะชวย confirm ผลที่เครื่องนับ การวินิจฉัยโรคนอกจากการใชขอมูลประวัติ การตรวจรางกาย เม็ดเลือดอัตโนมัติและยังเปนการชวยวินิจฉัยโรคไดงายและรวดเร็ว และการตรวจทางหองปฏิบัติการเบื้องตนไดแก CBC, Urine ศัพทตอไปนี้ใชในการบรรยายลักษณะตางๆ ของเม็ดเลือดแดง3 examination แลวการตรวจสเมียรเลือดจะทำาใหสามารถวินิจฉัย -