Hereditary Elliptocytosis CASE REPORT
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Tareq Al-Adaily Ibrahim Elhaj Ayah Fraihat Dana Alnasra Sheet
Anemia of decreased production II Sheet 3 – + Hemolytic anemia Dana Alnasra Ayah Fraihat Ibrahim Elhaj Tareq Al-adaily 0 **Flashback to previous lectures: − Anemia is the reduction of oxygen carrying capacity of blood secondary to a decrease in red cell mass. − We classified anemia according to cause into: 1. Anemia of blood loss (chronic and acute) 2. Anemia of decreased production 3. Hemolytic anemia − Then, we mentioned general causes for the anemia of decreased production, which are: nutritional deficiency, chronic inflammation, and bone marrow failure. We’ve already discussed the first two and now we will proceed to the last one. Anemias resulting from bone marrow failure: 1. Aplastic anemia 2. Pure red cell aplasia 3. Myelophthisic anemia 4. Myelodysplastic syndrome Other anemias of decreased production: 1. Anemia of renal failure 2. Anemia of liver disease 3. Anemia of hypothyroidism 00:00 1. Aplastic anemia Is a condition where the multipotent myeloid stem cells produced by the bone marrow are damaged. Remember: the bone marrow has stem cells called myeloid stem cells which eventually differentiate into erythrocytes, megakaryocytes (platelets), and myeloblasts (white blood cells). Notice that lymphocytes are not produced from the myeloid progenitor, therefore, they are not affected. depleted normal 1 | P a g e As a result, the bone marrow becomes depleted of hematopoietic cells. And this is reflected as peripheral blood pancytopenia (all blood cells -including reticulocytes- are decreased, with the exception of lymphocytes). Pathogenesis There are two forms of aplastic anemia according to pathogenesis: a. Acquired aplastic anemia It happens because of an extrinsic factor e.g. -

Hereditary Spherocytosis: Clinical Features
Title Overview: Hereditary Hematological Disorders of red cell shape. Disorders Red cell Enzyme disorders Disorders of Hemoglobin Inherited bleeding disorders- platelet disorders, coagulation factor Anthea Greenway MBBS FRACP FRCPA Visiting Associate deficiencies Division of Pediatric Hematology-Oncology Duke University Health Service Inherited Thrombophilia Hereditary Disorders of red cell Disorders of red cell shape (cytoskeleton): cytoskeleton: • Mutations of 5 proteins connect cytoskeleton of red cell to red cell membrane • Hereditary Spherocytosis- sphere – Spectrin (composed of alpha, beta heterodimers) –Ankyrin • Hereditary Elliptocytosis-ellipse, elongated forms – Pallidin (band 4.2) – Band 4.1 (protein 4.1) • Hereditary Pyropoikilocytosis-bizarre red cell forms – Band 3 protein (the anion exchanger, AE1) – RhAG (the Rh-associated glycoprotein) Normal red blood cell- discoid, with membrane flexibility Hereditary Spherocytosis: Clinical features: • Most common hereditary hemolytic disorder (red cell • Neonatal jaundice- severe (phototherapy), +/- anaemia membrane) • Hemolytic anemia- moderate in 60-75% cases • Mutations of one of 5 genes (chromosome 8) for • Severe hemolytic anaemia in 5% (AR, parents ASx) cytoskeletal proteins, overall effect is spectrin • fatigue, jaundice, dark urine deficiency, severity dependant on spectrin deficiency • SplenomegalSplenomegaly • 200-300:million births, most common in Northern • Chronic complications- growth impairment, gallstones European countries • Often follows clinical course of affected -
![Anormal Rbc in Peripheral Blood. [Repaired].Pdf](https://docslib.b-cdn.net/cover/4277/anormal-rbc-in-peripheral-blood-repaired-pdf-544277.webp)
Anormal Rbc in Peripheral Blood. [Repaired].Pdf
1. Acanthocyte 2. Burr-cell 3. Microcyte 1. Basophilic Normoblast 2. Polychromatic Normoblast 3. Pycnotic Normoblast 4. Plasmocyte 5. Eosinophil 6. Promyelocyte 1. Macrocyte 2. Elliptocyte 1. Microcyte 2. Normocyte 1. Polychromatic Erythrocyte 2. Acanthocyte 3. Elliptocyte 1. Polychromatic Normoblast 2. Pycnotic Normoblast 3. Neutrophil Myelocyte 4. Neutrophil Metamyelocyte 1. Schistocyte 2. Microcyte BASOPHILIC ( EARLY ) NORMOBLASTS Basophilic Erythroblast Basophilic Stippling, Blood smear, May-Giemsa stain, (×1000) CABOT'S RINGS Drepanocyte Elliptocyte Erythroblast ERYTHROBLAST in the blood Howell-jolly body Hypo chromic LACRYMOCYTES Leptocyte Malaria, Blood smear, May-Giemsa stain, ×1000 MICROCYTES Orthochromatic erythroblast Pappen heimer Bodies & 1. Schistocyte 2. Elliptocyte 3. Acanthocyte POIKILOCYTOSIS Polychromatic Erythroblast Pro Erytroblast Proerythroblasts Reticulocyte Rouleaux SICKLE CELLS Sickle cell Spherocyte Spherocyte Spherocyte SPHEROCYTES STOMATOCYTES Target Cells Tear Drop Cell, Blood smear, May-Giemsa stain, x1000 Anulocyte 1. Burr-cell 2. Elliptocyte 1. Macrocyte 2. Microcyte 3. Elliptocyte 4. Schistocyte 1. Ovalocyte 2. Lacrymocyte 3. Target cell 1. Polychromatic Erythrocyte 2. Basophilic Stippling 1. Proerythroblast 2. Basophilic Erythroblast 3. Intermediate Erythroblast 4. Late Erythroblast 5. Monocyte 6. Lymphocyte 1. Target-cell 2. Elliptocyte 3. Acanthocyte 4. Stomatocyte 5. Schistocyte 6. Polychromatophilic erythrocyte. 1.Pro erythroblast 2.Basophilic normoblast 3.Polychromatic normoblast 4.Pycnotic normoblast -

Alpha Thalassemia Trait
Alpha Thalassemia Trait Alpha Thalassemia Trait Produced by St. Jude Children’s Research Hospital, Departments of Hematology, Patient Education, 1 and Biomedical Communications. Funds were provided by St. Jude Children’s Research Hospital, ALSAC, and a grant from the Plough Foundation. This document is not intended to replace counseling by a trained health care professional or genetic counselor. Our aim is to promote active participation in your care and treatment by providing information and education. Questions about individual health concerns or specific treatment options should be discussed with your doctor. For general information on sickle cell disease and other blood disorders, please visit our Web site at www.stjude.org/sicklecell. Copyright © 2009 St. Jude Children’s Research Hospital Alpha thalassemia trait All red blood cells contain hemoglobin (HEE muh glow bin), which carries oxygen from your lungs to all parts of your body. Alpha thalassemia (thal uh SEE mee uh) trait is a condition that affects the amount of hemo- globin in the red blood cells. • Adult hemoglobin (hemoglobin A) is made of alpha and beta globins. • Normally, people have 4 genes for alpha globin with 2 genes on each chromosome (aa/aa). People with alpha thalassemia trait only have 2 genes for alpha globin, so their bodies make slightly less hemoglobin than normal. This trait was passed on from their parents, like hair color or eye color. A trait is different from a disease 2 Alpha thalassemia trait is not a disease. Normally, a trait will not make you sick. Parents who have alpha thalassemia trait can pass it on to their children. -

Methemoglobinemia and Ascorbate Deficiency in Hemoglobin E Β Thalassemia: Metabolic and Clinical Implications
From www.bloodjournal.org by guest on April 2, 2015. For personal use only. Plenary paper Methemoglobinemia and ascorbate deficiency in hemoglobin E  thalassemia: metabolic and clinical implications Angela Allen,1,2 Christopher Fisher,1 Anuja Premawardhena,3 Dayananda Bandara,4 Ashok Perera,4 Stephen Allen,2 Timothy St Pierre,5 Nancy Olivieri,6 and David Weatherall1 1MRC Molecular Haematology Unit, Weatherall Institute of Molecular Medicine, University of Oxford, John Radcliffe Hospital, Oxford, United Kingdom; 2College of Medicine, Swansea University, Swansea, United Kingdom; 3University of Kelaniya, Colombo, Sri Lanka; 4National Thalassaemia Centre, District Hospital, Kurunegala, Sri Lanka; 5School of Physics, University of Western Australia, Crawley, Australia; and 6Hemoglobinopathy Research, University Health Network, Toronto, ON During investigations of the phenotypic man hypoxia induction factor pathway is There was, in addition, a highly signifi- diversity of hemoglobin (Hb) E  thalasse- not totally dependent on ascorbate lev- cant correlation between methemoglobin mia, a patient was encountered with per- els. A follow-up study of 45 patients with levels, splenectomy, and factors that sistently high levels of methemoglobin HbE  thalassemia showed that methemo- modify the degree of globin-chain imbal- associated with a left-shift in the oxygen globin levels were significantly increased ance. Because methemoglobin levels are dissociation curve, profound ascorbate and that there was also a significant re- modified by several mechanisms and may deficiency, and clinical features of scurvy; duction in plasma ascorbate levels. Hap- play a role in both adaptation to anemia these abnormalities were corrected by toglobin levels were significantly re- and vascular damage, there is a strong treatment with vitamin C. -

Morphological Study of Human Blood for Different Diseases
Research Article ISSN: 2574 -1241 DOI: 10.26717/BJSTR.2020.30.004893 Morphological Study of Human Blood for Different Diseases Muzafar Shah1*, Haseena1, Kainat1, Noor Shaba1, Sania1, Sadia1, Akhtar Rasool2, Fazal Akbar2 and Muhammad Israr3 1Centre for Animal Sciences & Fisheries, University of Swat, Pakistan 2Centre for Biotechnology and Microbiology, University of Swat, Pakistan 3Department of Forensic Sciences, University of Swat, Pakistan *Corresponding author: Muzafar Shah, Centre for Animal Sciences & Fisheries, University of Swat, Pakistan ARTICLE INFO ABSTRACT Received: August 25, 2020 The aim of our study was the screening of blood cells on the basis of morphology for different diseased with Morphogenetic characters I e. ear lobe attachment, clinodactyly Published: September 07, 2020 and tongue rolling. For this purpose, 318 blood samples were collected randomly. Samples were examined under the compound microscopic by using 100x with standard Citation: Muzafar Shah, Haseena, method. The results show 63 samples were found normal while in 255 samples, different Kainat, Noor Shaba, Sania, Sadia, et al. types of morphological changes were observed which was 68.5%, in which Bite cell 36%, Morphological Study of Human Blood for Elliptocyte 34%, Tear drop cell 30%, Schistocyte 26%, Hypochromic cell 22.5%, Irregular Different Diseases. Biomed J Sci & Tech Res contracted cell 16%, Echinocytes 15.5%, Roleaux 8%, Boat shape 6.5%, Sickle cell 5%, Keratocyte 4% and Acanthocytes 1.5%. During the screening of slides, bite cell, elliptocyte, tear drop cell, schistocytes, hypochromic cell, irregular contracted cells were found 30(1)-2020.Keywords: BJSTR.Human MS.ID.004893. blood; Diseases; frequently while echinocytes, boat shape cell, acanthocytes, sickle cells and keratocytes Morphological; Acanthocytes; Keratocyte were found rarely. -
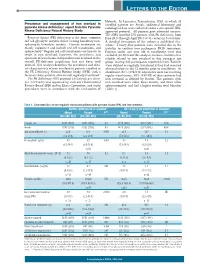
Prevalence and Management of Iron Overload in Pyruvate Kinase Deficiency
LETTERS TO THE EDITOR Helsinki. In Lancaster, Pennsylvania, USA, in which all Prevalence and management of iron overload in enrolled patients are Amish, additional laboratory and pyruvate kinase deficiency: report from the Pyruvate radiological data were collected under a site-specific IRB- Kinase Deficiency Natural History Study approved protocol. All patients gave informed consent. The NHS enrolled 278 patients with PK deficiency from Pyruvate kinase (PK) deficiency is the most common June 2014 through April 2017 at 31 centers in 6 countries. red cell glycolytic enzyme defect causing hereditary non- A detailed description of the cohort is published else- spherocytic hemolytic anemia. Current treatments are where.2 Twenty-four patients were excluded due to the mainly supportive and include red cell transfusions and inability to confirm two pathogenic PKLR mutations. splenectomy.1 Regular red cell transfusions are known to Patients under one year old at enrollment were also result in iron overload; however, the prevalence and excluded (n=12) from this analysis, because ferritin is less spectrum of transfusion-independent iron overload in the reliably related to iron overload in this youngest age overall PK-deficient population has not been well group, leaving 242 participants reported herein. Patients defined. This analysis describes the prevalence and clini- were defined as regularly transfused if they had received cal characteristics of iron overload in patients enrolled in ≥6 transfusions in the 12 months prior to enrollment. At the PK Deficiency Natural History Study (NHS) with a enrollment, 82% (198/242) of patients were not receiving focus on those patients who are not regularly transfused.2 regular transfusions; 38% (53/138) of these patients had The PK deficiency NHS protocol (clinicaltrials.gov identi- iron overload as defined by ferritin. -

Chapter 03- Diseases of the Blood and Certain Disorders Involving The
Chapter 3 Diseases of the blood and blood-forming organs and certain disorders involving the immune mechanism (D50- D89) Excludes2: autoimmune disease (systemic) NOS (M35.9) certain conditions originating in the perinatal period (P00-P96) complications of pregnancy, childbirth and the puerperium (O00-O9A) congenital malformations, deformations and chromosomal abnormalities (Q00-Q99) endocrine, nutritional and metabolic diseases (E00-E88) human immunodeficiency virus [HIV] disease (B20) injury, poisoning and certain other consequences of external causes (S00-T88) neoplasms (C00-D49) symptoms, signs and abnormal clinical and laboratory findings, not elsewhere classified (R00-R94) This chapter contains the following blocks: D50-D53 Nutritional anemias D55-D59 Hemolytic anemias D60-D64 Aplastic and other anemias and other bone marrow failure syndromes D65-D69 Coagulation defects, purpura and other hemorrhagic conditions D70-D77 Other disorders of blood and blood-forming organs D78 Intraoperative and postprocedural complications of the spleen D80-D89 Certain disorders involving the immune mechanism Nutritional anemias (D50-D53) D50 Iron deficiency anemia Includes: asiderotic anemia hypochromic anemia D50.0 Iron deficiency anemia secondary to blood loss (chronic) Posthemorrhagic anemia (chronic) Excludes1: acute posthemorrhagic anemia (D62) congenital anemia from fetal blood loss (P61.3) D50.1 Sideropenic dysphagia Kelly-Paterson syndrome Plummer-Vinson syndrome D50.8 Other iron deficiency anemias Iron deficiency anemia due to inadequate dietary -

Identifying Peripheral Blood Leukocytes and Erythrocytes in a Patient with Iron Deficiency Anemia
ADVANCED BLOOD CELL ID: IDENTIFYING PERIPHERAL BLOOD LEUKOCYTES AND ERYTHROCYTES IN A PATIENT WITH IRON DEFICIENCY ANEMIA Educational commentary is provided for participants enrolled in program #259- Advanced Blood Cell Identification. This virtual blood cell identification program includes case studies with more difficult challenges. To view the blood cell images in more detail, click on the sample identification numbers underlined in the paragraphs below. This will open a virtual image of the selected cell and the surrounding fields. If the image opens in the same window as the commentary, saving the commentary PDF and opening it outside your browser will allow you to switch between the commentary and the images more easily. Click on this link for the API ImageViewerTM Instructions. Learning Outcomes After completion of this exercise, participants will be able to: • describe morphologic features of monocytes and lymphocytes, and • identify distinguishing morphologic features in red blood cells associated with iron deficiency anemia. Case Study A 78 year old female patient was seen by her primary care physician due to extreme fatigue and headaches. The CBC results are as follows: WBC=9.3 x 109/L, RBC=4.43 x 1012/L, Hgb=8.7 g/dL, Hct=26.1%, MCV=58.9 fL, MCH=19.6 pg, MCHC=33.3 g/dL, RDW=24.8%, Platelet=425 x 109/L. Educational Commentary The cells annotated for commentary in this advanced testing event were selected from the peripheral blood smear of an elderly woman diagnosed with iron deficiency anemia (IDA). IDA is a common worldwide disorder. It can be caused by lack of adequate dietary iron, the malabsorption of iron, increased need for iron as in pregnancy or infancy and, most often, by bleeding. -

The Coexistence of Polycythemia Vera and Iron Deficiency Anemia Somchai Insiripong1, Wattana Insiripong2
CASE REPORT The Coexistence of Polycythemia Vera and Iron Deficiency Anemia Somchai Insiripong1, Wattana Insiripong2 1Department of Medicine, Saint Mary Hospital, Nakhon Ratchasima 30000, Thailand, 2Department of General Practice, NopparatRajathanee Hospital, Khanna Yao, Bangkok 10230, Thailand ABSTRACT Polycythemia vera (PV) is a clonal myeloproliferative neoplasm mainly characterized by an abnormal increase of erythroid precursor cells leading to increased red blood cells (RBC) production that is opposite to iron deficiency anemia (IDA) of which the RBC production is decreased due to iron deficiency. This report was aimed to present one patient who had coexistence of these two opposite entities of the RBC production. She was a 47-year-old Thai who was admitted because of acute coronary syndrome and she was accidentally found to have microcytosis of RBC despite normal hemoglobin (Hb) concentration, Hb 14.7 g%, mean corpuscular volume (MCV) 70.0 fL, white blood cells 12,400/mm3, and platelet 401,000/mm3. The Hb analysis showed only A2A, with normal Hb A2 percentage. The polymerase chain reaction for alpha thalassemia-1 genotype was tested negative. Due to neither alpha- nor beta-thalassemia trait detected, the iron study was performed: Serum ferritin 6.1 ng/mL, serum iron 64 ug/dl, and total iron binding capacity 198 ug/dl. The iron storage was seemingly insufficient; hence, iron supplement was started and continued for 4 months. Her blood tests showed: Hb 18.3 g%, MCV 87.2 fl, serum ferritin 31.7 ng/ml, erythropoietin <1 IU/l, positive JAK2 V617F mutation, and normal oxygen saturation. The diagnosis of PV was definitely concluded and she was finally treated with hydroxyurea and occasional phlebotomy. -

Congenital Methemoglobinemia Identified by Pulse Oximetry Screening Jennifer Ward, Jayashree Motwani, Nikki Baker, Matthew Nash, Andrew K
Congenital Methemoglobinemia Identified by Pulse Oximetry Screening Jennifer Ward, BMBS,a Jayashree Motwani, MBBS,b Nikki Baker, MSc,a Matthew Nash, MBChB,a Andrew K. Ewer, MD,a,c Gergely Toldi, MDa Congenital methemoglobinemia is a rare condition caused by cytochrome b5 abstract reductase deficiency, cytochrome b5 deficiency, or hemoglobin M disease. Newborn pulse oximetry screening was developed for the early detection of critical congenital heart disease; however, it also enables the early identification of other hypoxemic conditions. We present the case of a term neonate who was admitted to the neonatal unit after a failed pulse oximetry screening at 3 hours of age. Oxygen saturations remained between 89% and 92% despite an increase in oxygen therapy. Chest radiograph and echocardiogram results were normal. A capillary blood gas test had normal results except for a raised methemoglobin level of 16%. Improvement was Departments of aNeonatology and bHaematology, seen on the administration of methylene blue, which also resulted in an Birmingham Women’s and Children’s Hospital, Birmingham, increase in oxygen saturations to within normal limits. Further investigation United Kingdom; and cInstitute of Metabolism and Systems Research, University of Birmingham, Birmingham, United revealed evidence of type I hereditary cytochrome b5 reductase deficiency as Kingdom a result of a CYB5R3 gene mutation with 2 pathogenic variants involving Drs Ward and Toldi were responsible for neonatal guanine-to-adenine substitutions. Although mild cyanosis is generally the care and drafted and reviewed the manuscript; Dr only symptom of type I disease, patients may later develop associated Nash and Ms Baker were responsible for neonatal symptoms, such as fatigue and shortness of breath. -

Hemoglobin Bart's and Alpha Thalassemia Fact Sheet
Health Care Provider Hemoglobinopathy Fact Sheet Hemoglobin Bart’s & Alpha Thalassemia Hemoglobin Bart’s is a tetramer of gamma (fetal) globin chains seen during the newborn period. Its presence indicates that one or more of the four genes that produce alpha globin chains are dysfunctional, causing alpha thalassemia. The more alpha genes affected, the more significant the thalassemia and clinical symptoms. Alpha thalassemia occurs in individuals of all ethnic backgrounds and is one of the most common genetic diseases worldwide. However, the clinically significant forms (Hemoglobin H disease, Hemoglobin H Constant Spring, and Alpha Thalassemia Major) occur predominantly among Southeast Asians. Summarized below are the manifestations associated with the different levels of Hemoglobin Bart’s detected on the newborn screen, and recommendations for follow-up. The number of dysfunctional genes is estimated by the percentage of Bart’s seen on the newborn screen. Silent Carrier- Low Bart’s If only one alpha gene is affected, the other three genes can compensate nearly completely and only a low level of Bart’s is detected, unless hemoglobin Constant Spring is identified (see below). Levels of Bart’s below a certain percentage are not generally reported by the State Newborn Screening Program as these individuals are likely to be clinically and hematologically normal. However, a small number of babies reported as having possible alpha thalassemia trait will be silent carriers. Alpha Thalassemia or Hemoglobin Constant Spring Trait- Moderate Bart’s Alpha thalassemia trait produces a moderate level of Bart’s and typically results from the dysfunction of two alpha genes-- either due to gene deletions or a specific change in the alpha gene that produces elongated alpha globin and has a thalassemia-like effect: hemoglobin Constant Spring.