Somatic Increase of CCT8 Mimics Proteostasis of Human Pluripotent Stem Cells and Extends C. Elegans Lifespan
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-Like Mouse Models: Tracking the Role of the Hairless Gene
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 5-2006 Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene Yutao Liu University of Tennessee - Knoxville Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Part of the Life Sciences Commons Recommended Citation Liu, Yutao, "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino- like Mouse Models: Tracking the Role of the Hairless Gene. " PhD diss., University of Tennessee, 2006. https://trace.tennessee.edu/utk_graddiss/1824 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Yutao Liu entitled "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the requirements for the degree of Doctor of Philosophy, with a major in Life Sciences. Brynn H. Voy, Major Professor We have read this dissertation and recommend its acceptance: Naima Moustaid-Moussa, Yisong Wang, Rogert Hettich Accepted for the Council: Carolyn R. -
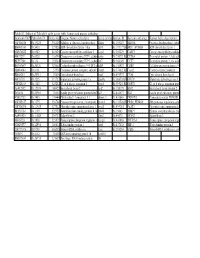
Table S3: Subset of Zebrafish Early Genes with Human And
Table S3: Subset of Zebrafish early genes with human and mouse orthologs Genbank ID(ZFZebrafish ID Entrez GenUnigene Name (zebrafish) Gene symbo Human ID Humann ortholog Human Gene description AW116838 Dr.19225 336425 Aldolase a, fructose-bisphosphate aldoa Hs.155247 ALDOA Fructose-bisphosphate aldola BM005100 Dr.5438 327026 ADP-ribosylation factor 1 like arf1l Hs.119177||HsARF1_HUMAN ADP-ribosylation factor 1 AW076882 Dr.6582 403025 Cancer susceptibility candidate 3 casc3 Hs.350229 CASC3 Cancer susceptibility candidat AI437239 Dr.6928 116994 Chaperonin containing TCP1, subun cct6a Hs.73072||Hs.CCT6A T-complex protein 1, zeta sub BE557308 Dr.134 192324 Chaperonin containing TCP1, subun cct7 Hs.368149 CCT7 T-complex protein 1, eta subu BG303647 Dr.26326 321602 Cyclin-dependent kinase 9 (CDC2-recdk9 Hs.150423 CDK9 Cell division protein kinase 9 AB040044 Dr.8161 57970 Coatomer protein complex, subunit zcopz1 Hs.37482||Hs.Copz2 Coatomer zeta-2 subunit BI888253 Dr.20911 30436 Eyes absent homolog 1 eya1 Hs.491997 EYA4 Eyes absent homolog 4 AI878758 Dr.3225 317737 Glutamate dehydrogenase 1a glud1a Hs.368538||HsGLUD1 Glutamate dehydrogenase 1, AW128619 Dr.1388 325284 G1 to S phase transition 1 gspt1 Hs.59523||Hs.GSPT1 G1 to S phase transition prote AF412832 Dr.12595 140427 Heat shock factor 2 hsf2 Hs.158195 HSF2 Heat shock factor protein 2 D38454 Dr.20916 30151 Insulin gene enhancer protein Islet3 isl3 Hs.444677 ISL2 Insulin gene enhancer protein AY052752 Dr.7485 170444 Pbx/knotted 1 homeobox 1.1 pknox1.1 Hs.431043 PKNOX1 Homeobox protein PKNOX1 -

A Rare Variant in MCF2L Identified Using Exclusion Linkage in A
European Journal of Human Genetics (2016) 24, 86–91 & 2016 Macmillan Publishers Limited All rights reserved 1018-4813/16 www.nature.com/ejhg ARTICLE A rare variant in MCF2L identified using exclusion linkage in a pedigree with premature atherosclerosis Stephanie Maiwald1,7, Mahdi M Motazacker1,7,8, Julian C van Capelleveen1, Suthesh Sivapalaratnam1, Allard C van der Wal2, Chris van der Loos2, John JP Kastelein1, Willem H Ouwehand3,4, G Kees Hovingh1, Mieke D Trip1,5, Jaap D van Buul6 and Geesje M Dallinga-Thie*,1 Cardiovascular disease (CVD) is a major cause of death in Western societies. CVD risk is largely genetically determined. The molecular pathology is, however, not elucidated in a large number of families suffering from CVD. We applied exclusion linkage analysis and next-generation sequencing to elucidate the molecular defect underlying premature CVD in a small pedigree, comprising two generations of which six members suffered from premature CVD. A total of three variants showed co-segregation with the disease status in the family. Two of these variants were excluded from further analysis based on the prevalence in replication cohorts, whereas a non-synonymous variant in MCF.2 Cell Line Derived Transforming Sequence-like protein (MCF2L, c.2066A4G; p.(Asp689Gly); NM_001112732.1), located in the DH domain, was only present in the studied family. MCF2L is a guanine exchange factor that potentially links pathways that signal through Rac1 and RhoA. Indeed, in HeLa cells, MCF2L689Gly failed to activate Rac1 as well as RhoA, resulting in impaired stress fiber formation. Moreover, MCF2L protein was found in human atherosclerotic lesions but not in healthy tissue segments. -

Supplemental Information
Supplemental information Dissection of the genomic structure of the miR-183/96/182 gene. Previously, we showed that the miR-183/96/182 cluster is an intergenic miRNA cluster, located in a ~60-kb interval between the genes encoding nuclear respiratory factor-1 (Nrf1) and ubiquitin-conjugating enzyme E2H (Ube2h) on mouse chr6qA3.3 (1). To start to uncover the genomic structure of the miR- 183/96/182 gene, we first studied genomic features around miR-183/96/182 in the UCSC genome browser (http://genome.UCSC.edu/), and identified two CpG islands 3.4-6.5 kb 5’ of pre-miR-183, the most 5’ miRNA of the cluster (Fig. 1A; Fig. S1 and Seq. S1). A cDNA clone, AK044220, located at 3.2-4.6 kb 5’ to pre-miR-183, encompasses the second CpG island (Fig. 1A; Fig. S1). We hypothesized that this cDNA clone was derived from 5’ exon(s) of the primary transcript of the miR-183/96/182 gene, as CpG islands are often associated with promoters (2). Supporting this hypothesis, multiple expressed sequences detected by gene-trap clones, including clone D016D06 (3, 4), were co-localized with the cDNA clone AK044220 (Fig. 1A; Fig. S1). Clone D016D06, deposited by the German GeneTrap Consortium (GGTC) (http://tikus.gsf.de) (3, 4), was derived from insertion of a retroviral construct, rFlpROSAβgeo in 129S2 ES cells (Fig. 1A and C). The rFlpROSAβgeo construct carries a promoterless reporter gene, the β−geo cassette - an in-frame fusion of the β-galactosidase and neomycin resistance (Neor) gene (5), with a splicing acceptor (SA) immediately upstream, and a polyA signal downstream of the β−geo cassette (Fig. -

CCT6A Sirna (Mouse)
For research purposes only, not for human use Product Data Sheet CCT6A siRNA (Mouse) Catalog # Source Reactivity Applications CRM0566 Synthetic M RNAi Description siRNA to inhibit CCT6A expression using RNA interference Specificity CCT6A siRNA (Mouse) is a target-specific 19-23 nt siRNA oligo duplexes designed to knock down gene expression. Form Lyophilized powder Gene Symbol CCT6A Alternative Names CCT6; CCTZ; CCTZ1; T-complex protein 1 subunit zeta; TCP-1-zeta; CCT-zeta-1 Entrez Gene 12466 (Mouse) SwissProt P80317 (Mouse) Purity > 97% Quality Control Oligonucleotide synthesis is monitored base by base through trityl analysis to ensure appropriate coupling efficiency. The oligo is subsequently purified by affinity-solid phase extraction. The annealed RNA duplex is further analyzed by mass spectrometry to verify the exact composition of the duplex. Each lot is compared to the previous lot by mass spectrometry to ensure maximum lot-to-lot consistency. Components We offers pre-designed sets of 3 different target-specific siRNA oligo duplexes of mouse CCT6A gene. Each vial contains 5 nmol of lyophilized siRNA. The duplexes can be transfected individually or pooled together to achieve knockdown of the target gene, which is most commonly assessed by qPCR or western blot. Our siRNA oligos are also chemically modified (2’-OMe) at no extra charge for increased stability and enhanced knockdown in vitro and in vivo. Directions for Use We recommends transfection with 100 nM siRNA 48 to 72 hours prior to cell lysis. Application key: E- ELISA, WB- -

Overexpressing CCT6A Contributes to Cancer Cell Growth by Affecting the G1-To-S Phase Transition and Predicts a Negative Prognosis in Hepatocellular Carcinoma
OncoTargets and Therapy Dovepress open access to scientific and medical research Open Access Full Text Article ORIGINAL RESEARCH Overexpressing CCT6A Contributes To Cancer Cell Growth By Affecting The G1-To-S Phase Transition And Predicts A Negative Prognosis In Hepatocellular Carcinoma This article was published in the following Dove Press journal: OncoTargets and Therapy Guofen Zeng1,* Purpose: To determine the oncogenic role of the sixth subunit of chaperonin-containing Jialiang Wang2,* tailless complex polypeptide 1 (CCT6A) in hepatocellular carcinoma (HCC) and address the Yanlin Huang 1,* correlation of CCT6A with clinicopathological characteristics and survival. Additionally, this Yifan Lian 2 study aimed to explore the effect of CCT6A on HCC cells and the underlying mechanisms. Dongmei Chen2 Methods: We searched for levels of CCT6A expression in the Oncomine database and GEPIA database, which was then validated by analyzing cancer and adjacent non-cancerous Huan Wei 2 tissues of HCC patients using quantitative PCR, Western blot, and immunohistochemistry Chaoshuang Lin1 1,2 assays. The relationship between CCT6A expression and survival was analyzed from the Yuehua Huang GEPIA database and confirmed by immunohistochemistry assays of 133 HCC tissue sec- 1Department of Infectious Diseases, The tions. In addition, the effect of depleting CCT6A on cell proliferation was assessed by CCK- fi Third Af liated Hospital of Sun Yat-sen 8 and colony formation assays. Cell cycle analysis, immunofluorescence assays, GSEA University, Guangzhou 510630, Guangdong, People’s Republic of China; analysis, and cyclin D expression analyzed by Western blot were used to explore the possible 2Guangdong Provincial Key Laboratory of underlying mechanism how dysregulated CCT6A affect the proliferation of HCC. -

Identification of Differentially Expressed Genes in Human Bladder Cancer Through Genome-Wide Gene Expression Profiling
521-531 24/7/06 18:28 Page 521 ONCOLOGY REPORTS 16: 521-531, 2006 521 Identification of differentially expressed genes in human bladder cancer through genome-wide gene expression profiling KAZUMORI KAWAKAMI1,3, HIDEKI ENOKIDA1, TOKUSHI TACHIWADA1, TAKENARI GOTANDA1, KENGO TSUNEYOSHI1, HIROYUKI KUBO1, KENRYU NISHIYAMA1, MASAKI TAKIGUCHI2, MASAYUKI NAKAGAWA1 and NAOHIKO SEKI3 1Department of Urology, Graduate School of Medical and Dental Sciences, Kagoshima University, 8-35-1 Sakuragaoka, Kagoshima 890-8520; Departments of 2Biochemistry and Genetics, and 3Functional Genomics, Graduate School of Medicine, Chiba University, 1-8-1 Inohana, Chuo-ku, Chiba 260-8670, Japan Received February 15, 2006; Accepted April 27, 2006 Abstract. Large-scale gene expression profiling is an effective CKS2 gene not only as a potential biomarker for diagnosing, strategy for understanding the progression of bladder cancer but also for staging human BC. This is the first report (BC). The aim of this study was to identify genes that are demonstrating that CKS2 expression is strongly correlated expressed differently in the course of BC progression and to with the progression of human BC. establish new biomarkers for BC. Specimens from 21 patients with pathologically confirmed superficial (n=10) or Introduction invasive (n=11) BC and 4 normal bladder samples were studied; samples from 14 of the 21 BC samples were subjected Bladder cancer (BC) is among the 5 most common to microarray analysis. The validity of the microarray results malignancies worldwide, and the 2nd most common tumor of was verified by real-time RT-PCR. Of the 136 up-regulated the genitourinary tract and the 2nd most common cause of genes we detected, 21 were present in all 14 BCs examined death in patients with cancer of the urinary tract (1-7). -

Open Full Page
Published OnlineFirst February 10, 2017; DOI: 10.1158/2159-8290.CD-16-1045 RESEARCH ARTICLE Interaction Landscape of Inherited Polymorphisms with Somatic Events in Cancer Hannah Carter 1 , 2 , 3 , 4 , Rachel Marty 5 , Matan Hofree 6 , Andrew M. Gross 5 , James Jensen 5 , Kathleen M. Fisch1,2,3,7 , Xingyu Wu 2 , Christopher DeBoever 5 , Eric L. Van Nostrand 4,8 , Yan Song 4,8 , Emily Wheeler 4,8 , Jason F. Kreisberg 1,3 , Scott M. Lippman 2 , Gene W. Yeo 4,8 , J. Silvio Gutkind 2 , 3 , and Trey Ideker 1 , 2 , 3 , 4 , 5,6 Downloaded from cancerdiscovery.aacrjournals.org on September 27, 2021. © 2017 American Association for Cancer Research. Published OnlineFirst February 10, 2017; DOI: 10.1158/2159-8290.CD-16-1045 ABSTRACT Recent studies have characterized the extensive somatic alterations that arise dur- ing cancer. However, the somatic evolution of a tumor may be signifi cantly affected by inherited polymorphisms carried in the germline. Here, we analyze genomic data for 5,954 tumors to reveal and systematically validate 412 genetic interactions between germline polymorphisms and major somatic events, including tumor formation in specifi c tissues and alteration of specifi c cancer genes. Among germline–somatic interactions, we found germline variants in RBFOX1 that increased incidence of SF3B1 somatic mutation by 8-fold via functional alterations in RNA splicing. Similarly, 19p13.3 variants were associated with a 4-fold increased likelihood of somatic mutations in PTEN. In support of this associ- ation, we found that PTEN knockdown sensitizes the MTOR pathway to high expression of the 19p13.3 gene GNA11 . -

A Survey of the Ce-Tubulin Gene Family in Chicken: Unexpected Sequence Heterogeneity in the Polypeptides Encoded by Five Expressed Genes
The EMBO Journal vol.7 no.4 pp.931 -940, 1988 A survey of the ce-tubulin gene family in chicken: unexpected sequence heterogeneity in the polypeptides encoded by five expressed genes Lisa F.Pratt1 and Don W.Cleveland encoding both a- and ,B-tubulin lay beneath this heterogeneity of microtubule utilization (see Cleveland and Sullivan, 1985; Department of Biological Chemistry, The Johns Hopkins University Cleveland, 1987). For ,3-tubulin, extensive analyses in School of Medicine, 725 N. Wolfe St., Baltimore, MD 21205, USA chicken (Sullivan and Cleveland, 1984; Sullivan et al., 1985, 'Present address: Division of Clinical Immunology, Scripps Clinic and 1986a,b; Murphy et al., 1987; Montiero and Cleveland, Research Foundation, 10666 N. Torrey Pines Road, La Jolla, CA 92037, USA 1988), mouse (Lewis et al., 1985a; Sullivan and Cleveland, 1986; Wang et al., 1986) and human (Hall et al., 1983; Lee Communicated by K.Weber et al., 1983; Lewis et al., 1985b; Sullivan and Cleveland, 1986) have revealed six functional vertebrate f-tubulins. At To characterize the a-tubulin gene family in chicken, we least four [and probably five-see Lopata and Cleveland have isolated five chicken a-tubulin genes and determined (1987)] of these represent distinct ,B-tubulin isotypes whose the majority of the sequences of the encoded polypep- sequences (particularly within a carboxy-terminal variable tides. Three of these (cA3, ca5/6 and ca8) encode novel, domain) have been evolutionarily conserved (Sullivan and expressed a-tubulins that have not previously been Cleveland, 1986; Wang et al., 1986). analyzed, whereas one gene segment is a pseudogene and For a-tubulin, the analyses are less complete. -

CCT6A Rabbit Pab
Leader in Biomolecular Solutions for Life Science CCT6A Rabbit pAb Catalog No.: A3589 1 Publications Basic Information Background Catalog No. The protein encoded by this gene is a molecular chaperone that is a member of the A3589 chaperonin containing TCP1 complex (CCT), also known as the TCP1 ring complex (TRiC). This complex consists of two identical stacked rings, each containing eight different Observed MW proteins. Unfolded polypeptides enter the central cavity of the complex and are folded 58kDa in an ATP-dependent manner. The complex folds various proteins, including actin and tubulin. Alternate transcriptional splice variants of this gene, encoding different Calculated MW isoforms, have been characterized. In addition, several pseudogenes of this gene have 53kDa/58kDa been located. Category Primary antibody Applications WB,IHC Cross-Reactivity Human, Mouse, Rat Recommended Dilutions Immunogen Information WB 1:500 - 1:2000 Gene ID Swiss Prot 908 P40227 IHC 1:50 - 1:100 Immunogen Recombinant fusion protein containing a sequence corresponding to amino acids 80-250 of human CCT6A (NP_001753.1). Synonyms CCT6A;CCT-zeta;CCT-zeta-1;CCT6;Cctz;HTR3;MoDP-2;TCP-1-zeta;TCP20;TCPZ;TTCP20 Contact Product Information www.abclonal.com Source Isotype Purification Rabbit IgG Affinity purification Storage Store at -20℃. Avoid freeze / thaw cycles. Buffer: PBS with 0.02% sodium azide,50% glycerol,pH7.3. Validation Data Western blot analysis of extracts of various cell lines, using CCT6A antibody (A3589) at 1:1000 dilution. Secondary antibody: HRP Goat Anti-Rabbit IgG (H+L) (AS014) at 1:10000 dilution. Lysates/proteins: 25ug per lane. Blocking buffer: 3% nonfat dry milk in TBST. -

Ultraconserved Elements Are Associated with Homeostatic Control of Splicing Regulators by Alternative Splicing and Nonsense-Mediated Decay
Downloaded from genesdev.cshlp.org on September 24, 2021 - Published by Cold Spring Harbor Laboratory Press Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay Julie Z. Ni,1 Leslie Grate,1 John Paul Donohue,1 Christine Preston,2 Naomi Nobida,2 Georgeann O’Brien,2 Lily Shiue,1 Tyson A. Clark,3 John E. Blume,3 and Manuel Ares Jr.1,2,4 1Center for Molecular Biology of RNA and Department of Molecular, Cell, and Developmental Biology, University of California at Santa Cruz, Santa Cruz, California 95064, USA; 2Hughes Undergraduate Research Laboratory, University of California at Santa Cruz, Santa Cruz, California 95064, USA; 3Affymetrix, Inc., Santa Clara, California 95051, USA Many alternative splicing events create RNAs with premature stop codons, suggesting that alternative splicing coupled with nonsense-mediated decay (AS-NMD) may regulate gene expression post-transcriptionally. We tested this idea in mice by blocking NMD and measuring changes in isoform representation using splicing-sensitive microarrays. We found a striking class of highly conserved stop codon-containing exons whose inclusion renders the transcript sensitive to NMD. A genomic search for additional examples identified >50 such exons in genes with a variety of functions. These exons are unusually frequent in genes that encode splicing activators and are unexpectedly enriched in the so-called “ultraconserved” elements in the mammalian lineage. Further analysis show that NMD of mRNAs for splicing activators such as SR proteins is triggered by splicing activation events, whereas NMD of the mRNAs for negatively acting hnRNP proteins is triggered by splicing repression, a polarity consistent with widespread homeostatic control of splicing regulator gene expression.