Novel Potential ALL Low-Risk Markers Revealed by Gene
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Inhibition of Breast and Brain Cancer Cell Growth by Bccipα, An
Oncogene (2001) 20, 336 ± 345 ã 2001 Nature Publishing Group All rights reserved 0950 ± 9232/01 $15.00 www.nature.com/onc Inhibition of breast and brain cancer cell growth by BCCIPa,an evolutionarily conserved nuclear protein that interacts with BRCA2 Jingmei Liu1, Yuan Yuan1,2, Juan Huan2 and Zhiyuan Shen*,1 1Department of Molecular Genetics and Microbiology, University of New Mexico Health Sciences Center; 915 Camino de Salud, NE. Albuquerque, New Mexico, NM 87131, USA; 2Graduate Program of Molecular Genetics, College of Medicine, University of Illinois at Chicago, 900 S. Ashland Ave. Chicago, Illinois, IL 60607, USA BRCA2 is a tumor suppressor gene involved in mammary mouse BRCA2. It is expected that important functions tumorigenesis. Although important functions have been of BRCA2 reside in these conserved domains. Based on assigned to a few conserved domains of BRCA2, little is the functional analysis of the conserved BRCA2 known about the longest internal conserved domain domains, several models have been proposed for the encoded by exons 14 ± 24. We identi®ed a novel protein, role of BRCA2 in tumor suppression. designated BCCIPa, that interacts with part of the An N-terminus conserved domain in exon 3 (amino internal conserved region of human BRCA2. Human acids 48 ± 105) has been implicated in transcriptional BCCIP represents a family of proteins that are regulation of gene expression (Milner et al., 1997; evolutionarily conserved, and contain three distinct Nordling et al., 1998). Deletion of this region has been domains: an N-terminus acidic domain (NAD) of 30 ± identi®ed in breast cancers (Nordling et al., 1998). -

Cytoplasmic Expression of Epithelial Cell Transforming Sequence 2 in Lung Adenocarcinoma and Its Implications for Malignant Progression
Laboratory Investigation (2019) 99:551–567 https://doi.org/10.1038/s41374-018-0142-4 ARTICLE Cytoplasmic expression of epithelial cell transforming sequence 2 in lung adenocarcinoma and its implications for malignant progression 1 2 3 1 2 2 Zeinab Kosibaty ● Yoshihiko Murata ● Yuko Minami ● Tomoko Dai ● Junko Kano ● Ryota Matsuoka ● 2 2 Noriyuki Nakano ● Masayuki Noguchi Received: 8 March 2018 / Revised: 14 August 2018 / Accepted: 20 August 2018 / Published online: 12 December 2018 © United States & Canadian Academy of Pathology 2018 Abstract Epithelial cell transforming sequence 2 (ECT2), a guanine nucleotide exchange factor, is predominantly localized in the nucleus of non-transformed cells and functions to regulate cytokinesis. ECT2 is also localized in the cytoplasm of cancer cells. Aberrant cytoplasmic expression of ECT2 is thought to drive tumor growth and invasion. In this study, we investigated the cytoplasmic expression of ECT2 and its prognostic and biological significance in lung adenocarcinoma. Western blotting of cellular fractions from the nucleus and cytoplasm was performed to determine the subcellular localization of ECT2 in lung adenocarcinoma cell lines. The cytoplasmic expression of ECT2 in 167 lung fi 1234567890();,: 1234567890();,: adenocarcinomas was evaluated by immunohistochemistry and its clinical signi cance was examined using Kaplan–Meier curves and Cox regression analysis. Scraping cytology specimens of 13 fresh lung adenocarcinomas were used to assess the subcellular localization of ECT2 and its phosphorylation at Thr790 (P-ECT2(T790)). We found that ECT2 was localized in both the nucleus and cytoplasm of lung adenocarcinoma cell lines and tumor tissues. Cytoplasmic expression of ECT2 was detected by immunohistochemistry in 83 (50%) of the lung adenocarcinomas, and was found to increase during cancer progression. -

Analysis of Trans Esnps Infers Regulatory Network Architecture
Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2014 © 2014 Anat Kreimer All rights reserved ABSTRACT Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer eSNPs are genetic variants associated with transcript expression levels. The characteristics of such variants highlight their importance and present a unique opportunity for studying gene regulation. eSNPs affect most genes and their cell type specificity can shed light on different processes that are activated in each cell. They can identify functional variants by connecting SNPs that are implicated in disease to a molecular mechanism. Examining eSNPs that are associated with distal genes can provide insights regarding the inference of regulatory networks but also presents challenges due to the high statistical burden of multiple testing. Such association studies allow: simultaneous investigation of many gene expression phenotypes without assuming any prior knowledge and identification of unknown regulators of gene expression while uncovering directionality. This thesis will focus on such distal eSNPs to map regulatory interactions between different loci and expose the architecture of the regulatory network defined by such interactions. We develop novel computational approaches and apply them to genetics-genomics data in human. We go beyond pairwise interactions to define network motifs, including regulatory modules and bi-fan structures, showing them to be prevalent in real data and exposing distinct attributes of such arrangements. We project eSNP associations onto a protein-protein interaction network to expose topological properties of eSNPs and their targets and highlight different modes of distal regulation. -

UCSD MOLECULE PAGES Doi:10.6072/H0.MP.A002549.01 Volume 1, Issue 2, 2012 Copyright UC Press, All Rights Reserved
UCSD MOLECULE PAGES doi:10.6072/H0.MP.A002549.01 Volume 1, Issue 2, 2012 Copyright UC Press, All rights reserved. Review Article Open Access WAVE2 Tadaomi Takenawa1, Shiro Suetsugu2, Daisuke Yamazaki3, Shusaku Kurisu1 WASP family verprolin-homologous protein 2 (WAVE2, also called WASF2) was originally identified by its sequence similarity at the carboxy-terminal VCA (verprolin, cofilin/central, acidic) domain with Wiskott-Aldrich syndrome protein (WASP) and N-WASP (neural WASP). In mammals, WAVE2 is ubiquitously expressed, and its two paralogs, WAVE1 (also called suppressor of cAMP receptor 1, SCAR1) and WAVE3, are predominantly expressed in the brain. The VCA domain of WASP and WAVE family proteins can activate the actin-related protein 2/3 (Arp2/3) complex, a major actin nucleator in cells. Proteins that can activate the Arp2/3 complex are now collectively known as nucleation-promoting factors (NPFs), and the WASP and WAVE families are a founding class of NPFs. The WAVE family has an amino-terminal WAVE homology domain (WHD domain, also called the SCAR homology domain, SHD) followed by the proline-rich region that interacts with various Src-homology 3 (SH3) domain proteins. The VCA domain located at the C-terminus. WAVE2, like WAVE1 and WAVE3, constitutively forms a huge heteropentameric protein complex (the WANP complex), binding through its WHD domain with Abi-1 (or its paralogs, Abi-2 and Abi-3), HSPC300 (also called Brick1), Nap1 (also called Hem-2 and NCKAP1), Sra1 (also called p140Sra1 and CYFIP1; its paralog is PIR121 or CYFIP2). The WANP complex is recruited to the plasma membrane by cooperative action of activated Rac GTPases and acidic phosphoinositides. -

Cell Division Symmetry Control and Cancer Stem Cells
AIMS Molecular Science, 7(2): 82–98. DOI: 10.3934/molsci.2020006 Received: 15 February 2020 Accepted: 26 April 2020 Published: 06 May 2020 http://www.aimspress.com/journal/Molecular Review Cell division symmetry control and cancer stem cells Sreemita Majumdar and Song-Tao Liu* Department of Biological Sciences, University of Toledo, Toledo, OH 43606, USA * Correspondence: Email: [email protected]; Tel: +14195307853. Table S1. Genes encoding polarity and fate-determinant proteins involved in asymmetric cell division. C. elegans1 D. melanogaster 1 Mammals1 Description2 Associated with/ Interactors 3 Cellular Localization (mammalian cell)4 Serine/threonine protein microtubule-associated protein cell membrane, peripheral and lateral, par-1 par-1 MARK1/2/3/4 kinase MAPT/TAU cytoplasm, dendrite RING, Lipid binding par-2 - - domain PDZ for membrane, cell junction, adherens junction, cell cortex, par-3 baz PARD3 Oligomerization domain at actin, PARD6 endomembrane system, NTD Continued on next page 2 C. elegans1 D. melanogaster 1 Mammals1 Description2 Associated with/ Interactors 3 Cellular Localization (mammalian cell)4 Serine/threonine-protein nucleus, mitochondria, cytoplasm, par-4 Lkb1 STK11/LKB1 STRAD complex kinase membrane 14-3-3 domain binding par-5 14-3-3 YWHAB phosphoserine/ adapter to many proteins cytoplasm phosphothreonine motif cell membrane, centriolar satellite, actin par-6 par-6 PARD6A/B/G PB1, CRIB, PDZ PARD3 cytoskeleton,centrosome, cytoplasm ,ruffles PARD3, and a PARD6 protein PB1, AGC-Kinase (PARD6A, PARD6B or PARD6G) pkc-3 aPKC PRKCI/Z domain, DAG binding, cytoplasm, nucleus, membrane and a GTPase protein (CDC42 or Zinc finger domain RAC1), LLGL1,ECT2 LRR and PDZ protein Cadherin, Scrib-APC-beta-catenin nucleoplasm, basolateral plasma membrane, let-413 scrib SCRIB family. -

The Rac Gtpase in Cancer: from Old Concepts to New Paradigms Marcelo G
Published OnlineFirst August 14, 2017; DOI: 10.1158/0008-5472.CAN-17-1456 Cancer Review Research The Rac GTPase in Cancer: From Old Concepts to New Paradigms Marcelo G. Kazanietz1 and Maria J. Caloca2 Abstract Rho family GTPases are critical regulators of cellular func- mislocalization of Rac signaling components. The unexpected tions that play important roles in cancer progression. Aberrant pro-oncogenic functions of Rac GTPase-activating proteins also activity of Rho small G-proteins, particularly Rac1 and their challenged the dogma that these negative Rac regulators solely regulators, is a hallmark of cancer and contributes to the act as tumor suppressors. The potential contribution of Rac tumorigenic and metastatic phenotypes of cancer cells. This hyperactivation to resistance to anticancer agents, including review examines the multiple mechanisms leading to Rac1 targeted therapies, as well as to the suppression of antitumor hyperactivation, particularly focusing on emerging paradigms immune response, highlights the critical need to develop ther- that involve gain-of-function mutations in Rac and guanine apeutic strategies to target the Rac pathway in a clinical setting. nucleotide exchange factors, defects in Rac1 degradation, and Cancer Res; 77(20); 5445–51. Ó2017 AACR. Introduction directed toward targeting Rho-regulated pathways for battling cancer. Exactly 25 years ago, two seminal papers by Alan Hall and Nearly all Rho GTPases act as molecular switches that cycle colleagues illuminated us with one of the most influential dis- between GDP-bound (inactive) and GTP-bound (active) forms. coveries in cancer signaling: the association of Ras-related small Activation is promoted by guanine nucleotide exchange factors GTPases of the Rho family with actin cytoskeleton reorganization (GEF) responsible for GDP dissociation, a process that normally (1, 2). -

Defining Functional Interactions During Biogenesis of Epithelial Junctions
ARTICLE Received 11 Dec 2015 | Accepted 13 Oct 2016 | Published 6 Dec 2016 | Updated 5 Jan 2017 DOI: 10.1038/ncomms13542 OPEN Defining functional interactions during biogenesis of epithelial junctions J.C. Erasmus1,*, S. Bruche1,*,w, L. Pizarro1,2,*, N. Maimari1,3,*, T. Poggioli1,w, C. Tomlinson4,J.Lees5, I. Zalivina1,w, A. Wheeler1,w, A. Alberts6, A. Russo2 & V.M.M. Braga1 In spite of extensive recent progress, a comprehensive understanding of how actin cytoskeleton remodelling supports stable junctions remains to be established. Here we design a platform that integrates actin functions with optimized phenotypic clustering and identify new cytoskeletal proteins, their functional hierarchy and pathways that modulate E-cadherin adhesion. Depletion of EEF1A, an actin bundling protein, increases E-cadherin levels at junctions without a corresponding reinforcement of cell–cell contacts. This unexpected result reflects a more dynamic and mobile junctional actin in EEF1A-depleted cells. A partner for EEF1A in cadherin contact maintenance is the formin DIAPH2, which interacts with EEF1A. In contrast, depletion of either the endocytic regulator TRIP10 or the Rho GTPase activator VAV2 reduces E-cadherin levels at junctions. TRIP10 binds to and requires VAV2 function for its junctional localization. Overall, we present new conceptual insights on junction stabilization, which integrate known and novel pathways with impact for epithelial morphogenesis, homeostasis and diseases. 1 National Heart and Lung Institute, Faculty of Medicine, Imperial College London, London SW7 2AZ, UK. 2 Computing Department, Imperial College London, London SW7 2AZ, UK. 3 Bioengineering Department, Faculty of Engineering, Imperial College London, London SW7 2AZ, UK. 4 Department of Surgery & Cancer, Faculty of Medicine, Imperial College London, London SW7 2AZ, UK. -

Systems Analysis Implicates WAVE2&Nbsp
JACC: BASIC TO TRANSLATIONAL SCIENCE VOL.5,NO.4,2020 ª 2020 THE AUTHORS. PUBLISHED BY ELSEVIER ON BEHALF OF THE AMERICAN COLLEGE OF CARDIOLOGY FOUNDATION. THIS IS AN OPEN ACCESS ARTICLE UNDER THE CC BY-NC-ND LICENSE (http://creativecommons.org/licenses/by-nc-nd/4.0/). PRECLINICAL RESEARCH Systems Analysis Implicates WAVE2 Complex in the Pathogenesis of Developmental Left-Sided Obstructive Heart Defects a b b b Jonathan J. Edwards, MD, Andrew D. Rouillard, PHD, Nicolas F. Fernandez, PHD, Zichen Wang, PHD, b c d d Alexander Lachmann, PHD, Sunita S. Shankaran, PHD, Brent W. Bisgrove, PHD, Bradley Demarest, MS, e f g h Nahid Turan, PHD, Deepak Srivastava, MD, Daniel Bernstein, MD, John Deanfield, MD, h i j k Alessandro Giardini, MD, PHD, George Porter, MD, PHD, Richard Kim, MD, Amy E. Roberts, MD, k l m m,n Jane W. Newburger, MD, MPH, Elizabeth Goldmuntz, MD, Martina Brueckner, MD, Richard P. Lifton, MD, PHD, o,p,q r,s t d Christine E. Seidman, MD, Wendy K. Chung, MD, PHD, Martin Tristani-Firouzi, MD, H. Joseph Yost, PHD, b u,v Avi Ma’ayan, PHD, Bruce D. Gelb, MD VISUAL ABSTRACT Edwards, J.J. et al. J Am Coll Cardiol Basic Trans Science. 2020;5(4):376–86. ISSN 2452-302X https://doi.org/10.1016/j.jacbts.2020.01.012 JACC: BASIC TO TRANSLATIONALSCIENCEVOL.5,NO.4,2020 Edwards et al. 377 APRIL 2020:376– 86 WAVE2 Complex in LVOTO HIGHLIGHTS ABBREVIATIONS AND ACRONYMS Combining CHD phenotype–driven gene set enrichment and CRISPR knockdown screening in zebrafish is an effective approach to identifying novel CHD genes. -

Statistical Methods for High-Dimensional Networked Data Analysis
Statistical Methods for High-Dimensional Networked Data Analysis by Yan Zhou A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Biostatistics) in The University of Michigan 2015 Doctoral Committee: Professor Peter X. K. Song, Chair Professor Matthias Kretzler Assistant Professor Xiaoquan William Wen Professor Ji Zhu c Yan Zhou 2015 All Rights Reserved To my family ii ACKNOWLEDGEMENTS I would like to acknowledge many people for their help throughout my graduate study. I would first like to express my deepest gratitude to my advisor Dr. Peter X.- K. Song, for sharing his knowledge, encouraging me to work hard and constantly try to improve my work, and giving me the freedom to explore a diverse set of projects. He is the one who first sparked my interest in networked data analysis. I am not only respectful for his immense knowledge, motivation, and enthusiasm towards research, but also appreciate his invaluable guidance and enthusiastic encouragement during many difficult moments in my research. I could not have imagined having a better advisor and mentor for my Ph.D. study. I would also like to thank my other thesis committee members Dr. Ji Zhu, Dr. Xiaoquan Willian Wen and Dr. Matthias Kretzler for their help and advices that made me more productive than what I could achieve on my own. All enlightening discussions I had with them have lead to better contents of this dissertation. In addition, I would like to acknowledge my collaborators, Dr. Pei Wang, Dr. Betsy Lozoff and Dr. Fengji Geng, who offered me excellent opportunities and con- structive suggestions to apply my statistical knowledge to solve real-world problems, which greatly strengthened the scientific background of my dissertation research. -
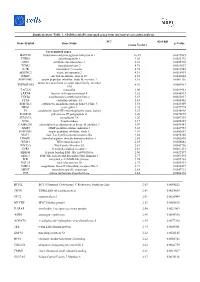
Supplementary Table 1. All Differentially Expressed Genes from Microarray Screening Analysis
Supplementary Table 1. All differentially expressed genes from microarray screening analysis. FCa (Gal-KD Gene Symbol Gene Name p-Value versus Vector) Up-regulated genes HAPLN1 hyaluronan and proteoglycan link protein 1 10.49 0.0027085 THBS1 thrombospondin 1 9.20 0.0022192 ODC1 ornithine decarboxylase 1 6.38 0.0055776 TGM2 transglutaminase 2 4.76 0.0015627 IL7R interleukin 7 receptor 4.75 0.0017245 SERINC2 serine incorporator 2 4.51 0.0014919 ITM2C integral membrane protein 2C 4.32 0.0044644 SERPINB7 serpin peptidase inhibitor, clade B, member 7 4.18 0.0081136 tumor necrosis factor receptor superfamily, member TNFRSF10D 4.01 0.0085561 10d TAGLN transgelin 3.90 0.0099963 LRRN4 leucine rich repeat neuronal 4 3.82 0.0046513 TGFB2 transforming growth factor beta 2 3.51 0.0035017 CPA4 carboxypeptidase A4 3.43 0.0008452 EPB41L3 erythrocyte membrane protein band 4.1-like 3 3.34 0.0025309 NRG1 neuregulin 1 3.28 0.0079724 F3 coagulation factor III (thromboplastin, tissue factor) 3.27 0.0038968 POLR3G polymerase III polypeptide G 3.26 0.0070675 SEMA7A semaphorin 7A 3.20 0.0087335 NT5E 5-nucleotidase 3.17 0.0036353 CAMK2N1 calmodulin-dependent protein kinase II inhibitor 1 3.07 0.0090141 TIMP3 TIMP metallopeptidase inhibitor 3 3.03 0.0047953 SERPINE1 serpin peptidase inhibitor, clade E 2.97 0.0053652 MALL mal, T-cell differentiation protein-like 2.88 0.0078205 DDAH1 dimethylarginine dimethylaminohydrolase 1 2.86 0.0002895 WDR3 WD repeat domain 3 2.85 0.0058842 WNT5A Wnt Family Member 5A 2.81 0.0043796 GPR1 G protein-coupled receptor 1 2.81 0.0021313 -

Effects and Mechanisms of Eps8 on the Biological Behaviour of Malignant Tumours (Review)
824 ONCOLOGY REPORTS 45: 824-834, 2021 Effects and mechanisms of Eps8 on the biological behaviour of malignant tumours (Review) KAILI LUO1, LEI ZHANG2, YUAN LIAO1, HONGYU ZHOU1, HONGYING YANG2, MIN LUO1 and CHEN QING1 1School of Pharmaceutical Sciences and Yunnan Key Laboratory of Pharmacology for Natural Products, Kunming Medical University, Kunming, Yunnan 650500; 2Department of Gynecology, Yunnan Tumor Hospital and The Third Affiliated Hospital of Kunming Medical University; Kunming, Yunnan 650118, P.R. China Received August 29, 2020; Accepted December 9, 2020 DOI: 10.3892/or.2021.7927 Abstract. Epidermal growth factor receptor pathway substrate 8 1. Introduction (Eps8) was initially identified as the substrate for the kinase activity of EGFR, improving the responsiveness of EGF, which Malignant tumours are uncontrolled cell proliferation diseases is involved in cell mitosis, differentiation and other physiological caused by oncogenes and ultimately lead to organ and body functions. Numerous studies over the last decade have demon- dysfunction (1). In recent decades, great progress has been strated that Eps8 is overexpressed in most ubiquitous malignant made in the study of genes and signalling pathways in tumours and subsequently binds with its receptor to activate tumorigenesis. Eps8 was identified by Fazioli et al in NIH-3T3 multiple signalling pathways. Eps8 not only participates in the murine fibroblasts via an approach that allows direct cloning regulation of malignant phenotypes, such as tumour proliferation, of intracellular substrates for receptor tyrosine kinases (RTKs) invasion, metastasis and drug resistance, but is also related to that was designed to study the EGFR signalling pathway. Eps8 the clinicopathological characteristics and prognosis of patients. -

Key Genes Regulating Skeletal Muscle Development and Growth in Farm Animals
animals Review Key Genes Regulating Skeletal Muscle Development and Growth in Farm Animals Mohammadreza Mohammadabadi 1 , Farhad Bordbar 1,* , Just Jensen 2 , Min Du 3 and Wei Guo 4 1 Department of Animal Science, Faculty of Agriculture, Shahid Bahonar University of Kerman, Kerman 77951, Iran; [email protected] 2 Center for Quantitative Genetics and Genomics, Aarhus University, 8210 Aarhus, Denmark; [email protected] 3 Washington Center for Muscle Biology, Department of Animal Sciences, Washington State University, Pullman, WA 99163, USA; [email protected] 4 Muscle Biology and Animal Biologics, Animal and Dairy Science, University of Wisconsin-Madison, Madison, WI 53558, USA; [email protected] * Correspondence: [email protected] Simple Summary: Skeletal muscle mass is an important economic trait, and muscle development and growth is a crucial factor to supply enough meat for human consumption. Thus, understanding (candidate) genes regulating skeletal muscle development is crucial for understanding molecular genetic regulation of muscle growth and can be benefit the meat industry toward the goal of in- creasing meat yields. During the past years, significant progress has been made for understanding these mechanisms, and thus, we decided to write a comprehensive review covering regulators and (candidate) genes crucial for muscle development and growth in farm animals. Detection of these genes and factors increases our understanding of muscle growth and development and is a great help for breeders to satisfy demands for meat production on a global scale. Citation: Mohammadabadi, M.; Abstract: Farm-animal species play crucial roles in satisfying demands for meat on a global scale, Bordbar, F.; Jensen, J.; Du, M.; Guo, W.