Conservation of the BRCA1 Gene
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Inhibition of Breast and Brain Cancer Cell Growth by Bccipα, An
Oncogene (2001) 20, 336 ± 345 ã 2001 Nature Publishing Group All rights reserved 0950 ± 9232/01 $15.00 www.nature.com/onc Inhibition of breast and brain cancer cell growth by BCCIPa,an evolutionarily conserved nuclear protein that interacts with BRCA2 Jingmei Liu1, Yuan Yuan1,2, Juan Huan2 and Zhiyuan Shen*,1 1Department of Molecular Genetics and Microbiology, University of New Mexico Health Sciences Center; 915 Camino de Salud, NE. Albuquerque, New Mexico, NM 87131, USA; 2Graduate Program of Molecular Genetics, College of Medicine, University of Illinois at Chicago, 900 S. Ashland Ave. Chicago, Illinois, IL 60607, USA BRCA2 is a tumor suppressor gene involved in mammary mouse BRCA2. It is expected that important functions tumorigenesis. Although important functions have been of BRCA2 reside in these conserved domains. Based on assigned to a few conserved domains of BRCA2, little is the functional analysis of the conserved BRCA2 known about the longest internal conserved domain domains, several models have been proposed for the encoded by exons 14 ± 24. We identi®ed a novel protein, role of BRCA2 in tumor suppression. designated BCCIPa, that interacts with part of the An N-terminus conserved domain in exon 3 (amino internal conserved region of human BRCA2. Human acids 48 ± 105) has been implicated in transcriptional BCCIP represents a family of proteins that are regulation of gene expression (Milner et al., 1997; evolutionarily conserved, and contain three distinct Nordling et al., 1998). Deletion of this region has been domains: an N-terminus acidic domain (NAD) of 30 ± identi®ed in breast cancers (Nordling et al., 1998). -

Analysis of Trans Esnps Infers Regulatory Network Architecture
Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2014 © 2014 Anat Kreimer All rights reserved ABSTRACT Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer eSNPs are genetic variants associated with transcript expression levels. The characteristics of such variants highlight their importance and present a unique opportunity for studying gene regulation. eSNPs affect most genes and their cell type specificity can shed light on different processes that are activated in each cell. They can identify functional variants by connecting SNPs that are implicated in disease to a molecular mechanism. Examining eSNPs that are associated with distal genes can provide insights regarding the inference of regulatory networks but also presents challenges due to the high statistical burden of multiple testing. Such association studies allow: simultaneous investigation of many gene expression phenotypes without assuming any prior knowledge and identification of unknown regulators of gene expression while uncovering directionality. This thesis will focus on such distal eSNPs to map regulatory interactions between different loci and expose the architecture of the regulatory network defined by such interactions. We develop novel computational approaches and apply them to genetics-genomics data in human. We go beyond pairwise interactions to define network motifs, including regulatory modules and bi-fan structures, showing them to be prevalent in real data and exposing distinct attributes of such arrangements. We project eSNP associations onto a protein-protein interaction network to expose topological properties of eSNPs and their targets and highlight different modes of distal regulation. -

Statistical Methods for High-Dimensional Networked Data Analysis
Statistical Methods for High-Dimensional Networked Data Analysis by Yan Zhou A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Biostatistics) in The University of Michigan 2015 Doctoral Committee: Professor Peter X. K. Song, Chair Professor Matthias Kretzler Assistant Professor Xiaoquan William Wen Professor Ji Zhu c Yan Zhou 2015 All Rights Reserved To my family ii ACKNOWLEDGEMENTS I would like to acknowledge many people for their help throughout my graduate study. I would first like to express my deepest gratitude to my advisor Dr. Peter X.- K. Song, for sharing his knowledge, encouraging me to work hard and constantly try to improve my work, and giving me the freedom to explore a diverse set of projects. He is the one who first sparked my interest in networked data analysis. I am not only respectful for his immense knowledge, motivation, and enthusiasm towards research, but also appreciate his invaluable guidance and enthusiastic encouragement during many difficult moments in my research. I could not have imagined having a better advisor and mentor for my Ph.D. study. I would also like to thank my other thesis committee members Dr. Ji Zhu, Dr. Xiaoquan Willian Wen and Dr. Matthias Kretzler for their help and advices that made me more productive than what I could achieve on my own. All enlightening discussions I had with them have lead to better contents of this dissertation. In addition, I would like to acknowledge my collaborators, Dr. Pei Wang, Dr. Betsy Lozoff and Dr. Fengji Geng, who offered me excellent opportunities and con- structive suggestions to apply my statistical knowledge to solve real-world problems, which greatly strengthened the scientific background of my dissertation research. -
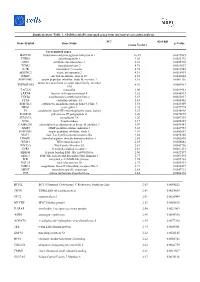
Supplementary Table 1. All Differentially Expressed Genes from Microarray Screening Analysis
Supplementary Table 1. All differentially expressed genes from microarray screening analysis. FCa (Gal-KD Gene Symbol Gene Name p-Value versus Vector) Up-regulated genes HAPLN1 hyaluronan and proteoglycan link protein 1 10.49 0.0027085 THBS1 thrombospondin 1 9.20 0.0022192 ODC1 ornithine decarboxylase 1 6.38 0.0055776 TGM2 transglutaminase 2 4.76 0.0015627 IL7R interleukin 7 receptor 4.75 0.0017245 SERINC2 serine incorporator 2 4.51 0.0014919 ITM2C integral membrane protein 2C 4.32 0.0044644 SERPINB7 serpin peptidase inhibitor, clade B, member 7 4.18 0.0081136 tumor necrosis factor receptor superfamily, member TNFRSF10D 4.01 0.0085561 10d TAGLN transgelin 3.90 0.0099963 LRRN4 leucine rich repeat neuronal 4 3.82 0.0046513 TGFB2 transforming growth factor beta 2 3.51 0.0035017 CPA4 carboxypeptidase A4 3.43 0.0008452 EPB41L3 erythrocyte membrane protein band 4.1-like 3 3.34 0.0025309 NRG1 neuregulin 1 3.28 0.0079724 F3 coagulation factor III (thromboplastin, tissue factor) 3.27 0.0038968 POLR3G polymerase III polypeptide G 3.26 0.0070675 SEMA7A semaphorin 7A 3.20 0.0087335 NT5E 5-nucleotidase 3.17 0.0036353 CAMK2N1 calmodulin-dependent protein kinase II inhibitor 1 3.07 0.0090141 TIMP3 TIMP metallopeptidase inhibitor 3 3.03 0.0047953 SERPINE1 serpin peptidase inhibitor, clade E 2.97 0.0053652 MALL mal, T-cell differentiation protein-like 2.88 0.0078205 DDAH1 dimethylarginine dimethylaminohydrolase 1 2.86 0.0002895 WDR3 WD repeat domain 3 2.85 0.0058842 WNT5A Wnt Family Member 5A 2.81 0.0043796 GPR1 G protein-coupled receptor 1 2.81 0.0021313 -

Efficient Zygotic Genome Editing Via RAD51-Enhanced Interhomolog Repair 2 3 Jonathan J
bioRxiv preprint doi: https://doi.org/10.1101/263699; this version posted August 6, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Efficient Zygotic Genome Editing via RAD51-Enhanced Interhomolog Repair 2 3 Jonathan J. Wilde1,3, Tomomi Aida1,3, Martin Wienisch1, Qiangge Zhang1, Peimin Qi1, and 4 Guoping Feng1,2* 5 6 Affiliations: 7 1McGovern Institute for Brain Research, Department of Brain and Cognitive Sciences, 8 Massachusetts Institute of Technology (MIT), Cambridge, Massachusetts 02139, USA 9 10 2Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, 11 Massachusetts 02142, USA 12 13 3These authors contributed equally to this work. 14 15 *E-mail: [email protected] 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 bioRxiv preprint doi: https://doi.org/10.1101/263699; this version posted August 6, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 47 Recent advances in genome editing have greatly improved knock-in (KI) efficiency1- 48 9. Searching for factors to further improve KI efficiency for therapeutic use and generation 49 of non-human primate (NHP) models, we found that the strand exchange protein RAD51 50 can significantly increase homozygous KI using CRISPR/Cas9 in mouse embryos through 51 an interhomolog repair (IHR) mechanism. -

Downloaded the “Top Edge” Version
bioRxiv preprint doi: https://doi.org/10.1101/855338; this version posted December 6, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 Drosophila models of pathogenic copy-number variant genes show global and 2 non-neuronal defects during development 3 Short title: Non-neuronal defects of fly homologs of CNV genes 4 Tanzeen Yusuff1,4, Matthew Jensen1,4, Sneha Yennawar1,4, Lucilla Pizzo1, Siddharth 5 Karthikeyan1, Dagny J. Gould1, Avik Sarker1, Yurika Matsui1,2, Janani Iyer1, Zhi-Chun Lai1,2, 6 and Santhosh Girirajan1,3* 7 8 1. Department of Biochemistry and Molecular Biology, Pennsylvania State University, 9 University Park, PA 16802 10 2. Department of Biology, Pennsylvania State University, University Park, PA 16802 11 3. Department of Anthropology, Pennsylvania State University, University Park, PA 16802 12 4 contributed equally to work 13 14 *Correspondence: 15 Santhosh Girirajan, MBBS, PhD 16 205A Life Sciences Building 17 Pennsylvania State University 18 University Park, PA 16802 19 E-mail: [email protected] 20 Phone: 814-865-0674 21 1 bioRxiv preprint doi: https://doi.org/10.1101/855338; this version posted December 6, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 22 ABSTRACT 23 While rare pathogenic copy-number variants (CNVs) are associated with both neuronal and non- 24 neuronal phenotypes, functional studies evaluating these regions have focused on the molecular 25 basis of neuronal defects. -

YY1/BCCIP Coordinately Regulates P53-Responsive Element (P53re)-Mediated Transactivation of P21waf1/Cip1
International Journal of Molecular Sciences Article YY1/BCCIP Coordinately Regulates P53-Responsive Element (p53RE)-Mediated Transactivation of p21Waf1/Cip1 Yi Sui 1, Tingting Wu 1, Fuqiang Li 2, Fei Wang 1, Yong Cai 1,3,4,* and Jingji Jin 1,3,4,* 1 Department of Biochemistry and Molecular Biology, School of Life Sciences, Jilin University, Changchun 130012, China; [email protected] (Y.S.); [email protected] (T.W.); [email protected] (F.W.) 2 School of Pharmacy, Changchun University of Chinese Medicine, Changchun 130117, China; [email protected] 3 National Engineering Laboratory for AIDS Vaccine, School of Life Sciences, Jilin University, Changchun 130012, China 4 Key Laboratory for Molecular Enzymology and Engineering, the Ministry of Education, Jilin University, Changchun 130012, China * Correspondence: [email protected] (Y.C.); [email protected] (J.J.); Tel.: +86-431-8515-5132 (Y.C.); +86-431-8515-5475 (J.J.) Received: 1 April 2019; Accepted: 26 April 2019; Published: 28 April 2019 Abstract: Transactivation of p21 (cyclin-dependent kinase inhibitor 1A, CDKN1A) is closely related to the recruitment of transcription cofactors at the p53 responsive elements (p53REs) in its promoter region. Human chromatin remodeling enzyme INO80 can be recruited to the p53REs of p21 promoter and negatively regulates p21. As one of the key subunits of the INO80 complex, YY1 has also been confirmed to bind to the p53RE sites of p21 promoter. Importantly, YY1 was recently reported to be bound and stabilized by BCCIP (BRCA2 and CDKN1A-interacting protein). Therefore, we hypothesized that the YY1/BCCIP complex plays an important role in regulating the transactivation of p21. -

Genetic Alterations of Protein Tyrosine Phosphatases in Human Cancers
Oncogene (2015) 34, 3885–3894 © 2015 Macmillan Publishers Limited All rights reserved 0950-9232/15 www.nature.com/onc REVIEW Genetic alterations of protein tyrosine phosphatases in human cancers S Zhao1,2,3, D Sedwick3,4 and Z Wang2,3 Protein tyrosine phosphatases (PTPs) are enzymes that remove phosphate from tyrosine residues in proteins. Recent whole-exome sequencing of human cancer genomes reveals that many PTPs are frequently mutated in a variety of cancers. Among these mutated PTPs, PTP receptor T (PTPRT) appears to be the most frequently mutated PTP in human cancers. Beside PTPN11, which functions as an oncogene in leukemia, genetic and functional studies indicate that most of mutant PTPs are tumor suppressor genes. Identification of the substrates and corresponding kinases of the mutant PTPs may provide novel therapeutic targets for cancers harboring these mutant PTPs. Oncogene (2015) 34, 3885–3894; doi:10.1038/onc.2014.326; published online 29 September 2014 INTRODUCTION tyrosine/threonine-specific phosphatases. (4) Class IV PTPs include Protein tyrosine phosphorylation has a critical role in virtually all four Drosophila Eya homologs (Eya1, Eya2, Eya3 and Eya4), which human cellular processes that are involved in oncogenesis.1 can dephosphorylate both tyrosine and serine residues. Protein tyrosine phosphorylation is coordinately regulated by protein tyrosine kinases (PTKs) and protein tyrosine phosphatases 1 THE THREE-DIMENSIONAL STRUCTURE AND CATALYTIC (PTPs). Although PTKs add phosphate to tyrosine residues in MECHANISM OF PTPS proteins, PTPs remove it. Many PTKs are well-documented oncogenes.1 Recent cancer genomic studies provided compelling The three-dimensional structures of the catalytic domains of evidence that many PTPs function as tumor suppressor genes, classical PTPs (RPTPs and non-RPTPs) are extremely well because a majority of PTP mutations that have been identified in conserved.5 Even the catalytic domain structures of the dual- human cancers are loss-of-function mutations. -

Enhancer Rnas: Transcriptional Regulators and Workmates of Namirnas in Myogenesis
Odame et al. Cell Mol Biol Lett (2021) 26:4 https://doi.org/10.1186/s11658-021-00248-x Cellular & Molecular Biology Letters REVIEW Open Access Enhancer RNAs: transcriptional regulators and workmates of NamiRNAs in myogenesis Emmanuel Odame , Yuan Chen, Shuailong Zheng, Dinghui Dai, Bismark Kyei, Siyuan Zhan, Jiaxue Cao, Jiazhong Guo, Tao Zhong, Linjie Wang, Li Li* and Hongping Zhang* *Correspondence: [email protected]; zhp@sicau. Abstract edu.cn miRNAs are well known to be gene repressors. A newly identifed class of miRNAs Farm Animal Genetic Resources Exploration termed nuclear activating miRNAs (NamiRNAs), transcribed from miRNA loci that and Innovation Key exhibit enhancer features, promote gene expression via binding to the promoter and Laboratory of Sichuan enhancer marker regions of the target genes. Meanwhile, activated enhancers pro- Province, College of Animal Science and Technology, duce endogenous non-coding RNAs (named enhancer RNAs, eRNAs) to activate gene Sichuan Agricultural expression. During chromatin looping, transcribed eRNAs interact with NamiRNAs University, Chengdu 611130, through enhancer-promoter interaction to perform similar functions. Here, we review China the functional diferences and similarities between eRNAs and NamiRNAs in myogen- esis and disease. We also propose models demonstrating their mutual mechanism and function. We conclude that eRNAs are active molecules, transcriptional regulators, and partners of NamiRNAs, rather than mere RNAs produced during enhancer activation. Keywords: Enhancer RNA, NamiRNAs, MicroRNA, Myogenesis, Transcriptional regulator Introduction Te identifcation of lin-4 miRNA in Caenorhabditis elegans in 1993 [1] triggered research to discover and understand small microRNAs’ (miRNAs) mechanisms. Recently, some miRNAs are reported to activate target genes during transcription via base pairing to the 3ʹ or 5ʹ untranslated regions (3ʹ or 5ʹ UTRs), the promoter [2], and the enhancer regions [3]. -

Novel Potential ALL Low-Risk Markers Revealed by Gene
Leukemia (2003) 17, 1891–1900 & 2003 Nature Publishing Group All rights reserved 0887-6924/03 $25.00 www.nature.com/leu BIO-TECHNICAL METHODS (BTS) Novel potential ALL low-risk markers revealed by gene expression profiling with new high-throughput SSH–CCS–PCR J Qiu1,5, P Gunaratne2,5, LE Peterson3, D Khurana2, N Walsham2, H Loulseged2, RJ Karni1, E Roussel4, RA Gibbs2, JF Margolin1,6 and MC Gingras1,6 1Texas Children’s Cancer Center and Department of Pediatrics; 2Human Genome Sequencing Center, Department of Molecular and Human Genetics; 3Department of Internal Medicine; 1,2,3 are all departments of Baylor College of Medicine, Baylor College of Medicine, Houston, TX, USA; and 4BioTher Corporation, Houston, TX, USA The current systems of risk grouping in pediatric acute t(1;19), BCR-ABL t(9;22), and MLL-AF4 t(4;11).1 These chromo- lymphoblastic leukemia (ALL) fail to predict therapeutic suc- somal modifications and other clinical findings such as age and cess in 10–35% of patients. To identify better predictive markers of clinical behavior in ALL, we have developed an integrated initial white blood cell count (WBC) define pediatric ALL approach for gene expression profiling that couples suppres- subgroups and are used as diagnostic and prognostic markers to sion subtractive hybridization, concatenated cDNA sequencing, assign specific risk-adjusted therapies. For instance, 1.0 to 9.9- and reverse transcriptase real-time quantitative PCR. Using this year-old patients with none of the determinant chromosomal approach, a total of 600 differentially expressed genes were translocation (NDCT) mentioned above but with a WBC higher identified between t(4;11) ALL and pre-B ALL with no determi- than 50 000 cells/ml are associated with higher risk group.2 nant chromosomal translocation. -

Essential Roles of BCCIP in Mouse Embryonic Development and Structural Stability of Chromosomes
Essential Roles of BCCIP in Mouse Embryonic Development and Structural Stability of Chromosomes Huimei Lu1,2, Yi-Yuan Huang1,2, Sonam Mehrotra1,2, Roberto Droz-Rosario1,2, Jingmei Liu1,2, Mantu Bhaumik1,3, Eileen White1,4, Zhiyuan Shen1,2* 1 The Cancer Institute of New Jersey, Robert Wood Johnson Medical School, University of Medicine and Dentistry of New Jersey, New Brunswick, New Jersey, United States of America, 2 Department of Radiation Oncology, Robert Wood Johnson Medical School, University of Medicine and Dentistry of New Jersey, New Brunswick, New Jersey, United States of America, 3 Department of Pediatrics, Robert Wood Johnson Medical School, University of Medicine and Dentistry of New Jersey, New Brunswick, New Jersey, United States of America, 4 Department of Molecular Biology and Biochemistry, Rutgers – The State University of New Jersey, Piscataway, New Jersey, United States of America Abstract BCCIP is a BRCA2- and CDKN1A(p21)-interacting protein that has been implicated in the maintenance of genomic integrity. To understand the in vivo functions of BCCIP, we generated a conditional BCCIP knockdown transgenic mouse model using Cre-LoxP mediated RNA interference. The BCCIP knockdown embryos displayed impaired cellular proliferation and apoptosis at day E7.5. Consistent with these results, the in vitro proliferation of blastocysts and mouse embryonic fibroblasts (MEFs) of BCCIP knockdown mice were impaired considerably. The BCCIP deficient mouse embryos die before E11.5 day. Deletion of the p53 gene could not rescue the embryonic lethality due to BCCIP deficiency, but partially rescues the growth delay of mouse embryonic fibroblasts in vitro. To further understand the cause of development and proliferation defects in BCCIP-deficient mice, MEFs were subjected to chromosome stability analysis. -

Gene Expression and Functionality in Head and Neck Cancer
ONCOLOGY REPORTS 35: 2431-2440, 2016 Spheroid-based 3-dimensional culture models: Gene expression and functionality in head and neck cancer MARIANNE SCHMIDT1, CLAUS-JUERGEN SCHOLZ2, CHRISTINE POLEDNIK1 and JEANETTE ROLLER1 1Department of Otorhinolaryngology, University of Würzburg, D-97080 Würzburg; 2Interdisciplinary Center for Clinical Research, Microarray Core Unit, University of Würzburg, D-97078 Würzburg, Germany Received October 26, 2015; Accepted December 5, 2015 DOI: 10.3892/or.2016.4581 Abstract. In the present study a panel of 12 head and neck Introduction cancer (HNSCC) cell lines were tested for spheroid forma- tion. Since the size and morphology of spheroids is dependent According to an analysis of over 3,000 cases of primary head on both cell adhesion and proliferation in the 3-dimensional and neck tumours in Germany in 2009, the patient benefit has (3D) context, morphology of HNSCC spheroids was related to not significantly improved from 1995 to 2006, despite new expression of E-cadherin and the proliferation marker Ki67. In treatment strategies. Particularly, the 5-year overall survival HNSCC cell lines the formation of tight regular spheroids was rate for carcinomas of hypopharyngeal origin is very low with dependent on distinct E-cadherin expression levels in mono- 27.2% (1). Hypopharyngeal cancer is often detected in later layer cultures, usually resulting in upregulation following stages, since early signs and symptoms rarely occur. Late aggregation into 3D structures. Cell lines expressing only low stages frequently involve multiple node affection, leading levels of E-cadherin in monolayers produced only loose cell to poor prognosis (http://emedicine.medscape.com/article clusters, frequently decreasing E-cadherin expression further /1375268).