Enhancer Rnas: Transcriptional Regulators and Workmates of Namirnas in Myogenesis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Inhibition of Breast and Brain Cancer Cell Growth by Bccipα, An
Oncogene (2001) 20, 336 ± 345 ã 2001 Nature Publishing Group All rights reserved 0950 ± 9232/01 $15.00 www.nature.com/onc Inhibition of breast and brain cancer cell growth by BCCIPa,an evolutionarily conserved nuclear protein that interacts with BRCA2 Jingmei Liu1, Yuan Yuan1,2, Juan Huan2 and Zhiyuan Shen*,1 1Department of Molecular Genetics and Microbiology, University of New Mexico Health Sciences Center; 915 Camino de Salud, NE. Albuquerque, New Mexico, NM 87131, USA; 2Graduate Program of Molecular Genetics, College of Medicine, University of Illinois at Chicago, 900 S. Ashland Ave. Chicago, Illinois, IL 60607, USA BRCA2 is a tumor suppressor gene involved in mammary mouse BRCA2. It is expected that important functions tumorigenesis. Although important functions have been of BRCA2 reside in these conserved domains. Based on assigned to a few conserved domains of BRCA2, little is the functional analysis of the conserved BRCA2 known about the longest internal conserved domain domains, several models have been proposed for the encoded by exons 14 ± 24. We identi®ed a novel protein, role of BRCA2 in tumor suppression. designated BCCIPa, that interacts with part of the An N-terminus conserved domain in exon 3 (amino internal conserved region of human BRCA2. Human acids 48 ± 105) has been implicated in transcriptional BCCIP represents a family of proteins that are regulation of gene expression (Milner et al., 1997; evolutionarily conserved, and contain three distinct Nordling et al., 1998). Deletion of this region has been domains: an N-terminus acidic domain (NAD) of 30 ± identi®ed in breast cancers (Nordling et al., 1998). -

PARSANA-DISSERTATION-2020.Pdf
DECIPHERING TRANSCRIPTIONAL PATTERNS OF GENE REGULATION: A COMPUTATIONAL APPROACH by Princy Parsana A dissertation submitted to The Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland July, 2020 © 2020 Princy Parsana All rights reserved Abstract With rapid advancements in sequencing technology, we now have the ability to sequence the entire human genome, and to quantify expression of tens of thousands of genes from hundreds of individuals. This provides an extraordinary opportunity to learn phenotype relevant genomic patterns that can improve our understanding of molecular and cellular processes underlying a trait. The high dimensional nature of genomic data presents a range of computational and statistical challenges. This dissertation presents a compilation of projects that were driven by the motivation to efficiently capture gene regulatory patterns in the human transcriptome, while addressing statistical and computational challenges that accompany this data. We attempt to address two major difficulties in this domain: a) artifacts and noise in transcriptomic data, andb) limited statistical power. First, we present our work on investigating the effect of artifactual variation in gene expression data and its impact on trans-eQTL discovery. Here we performed an in-depth analysis of diverse pre-recorded covariates and latent confounders to understand their contribution to heterogeneity in gene expression measurements. Next, we discovered 673 trans-eQTLs across 16 human tissues using v6 data from the Genotype Tissue Expression (GTEx) project. Finally, we characterized two trait-associated trans-eQTLs; one in Skeletal Muscle and another in Thyroid. Second, we present a principal component based residualization method to correct gene expression measurements prior to reconstruction of co-expression networks. -

Gene Expression Polarization
Transcriptional Profiling of the Human Monocyte-to-Macrophage Differentiation and Polarization: New Molecules and Patterns of Gene Expression This information is current as of September 27, 2021. Fernando O. Martinez, Siamon Gordon, Massimo Locati and Alberto Mantovani J Immunol 2006; 177:7303-7311; ; doi: 10.4049/jimmunol.177.10.7303 http://www.jimmunol.org/content/177/10/7303 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2006/11/03/177.10.7303.DC1 Material http://www.jimmunol.org/ References This article cites 61 articles, 22 of which you can access for free at: http://www.jimmunol.org/content/177/10/7303.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision by guest on September 27, 2021 • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2006 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Transcriptional Profiling of the Human Monocyte-to-Macrophage Differentiation and Polarization: New Molecules and Patterns of Gene Expression1 Fernando O. -

Analysis of Trans Esnps Infers Regulatory Network Architecture
Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2014 © 2014 Anat Kreimer All rights reserved ABSTRACT Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer eSNPs are genetic variants associated with transcript expression levels. The characteristics of such variants highlight their importance and present a unique opportunity for studying gene regulation. eSNPs affect most genes and their cell type specificity can shed light on different processes that are activated in each cell. They can identify functional variants by connecting SNPs that are implicated in disease to a molecular mechanism. Examining eSNPs that are associated with distal genes can provide insights regarding the inference of regulatory networks but also presents challenges due to the high statistical burden of multiple testing. Such association studies allow: simultaneous investigation of many gene expression phenotypes without assuming any prior knowledge and identification of unknown regulators of gene expression while uncovering directionality. This thesis will focus on such distal eSNPs to map regulatory interactions between different loci and expose the architecture of the regulatory network defined by such interactions. We develop novel computational approaches and apply them to genetics-genomics data in human. We go beyond pairwise interactions to define network motifs, including regulatory modules and bi-fan structures, showing them to be prevalent in real data and exposing distinct attributes of such arrangements. We project eSNP associations onto a protein-protein interaction network to expose topological properties of eSNPs and their targets and highlight different modes of distal regulation. -

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

The Expression of the Human Apolipoprotein Genes and Their Regulation by Ppars
CORE Metadata, citation and similar papers at core.ac.uk Provided by UEF Electronic Publications The expression of the human apolipoprotein genes and their regulation by PPARs Juuso Uski M.Sc. Thesis Biochemistry Department of Biosciences University of Kuopio June 2008 Abstract The expression of the human apolipoprotein genes and their regulation by PPARs. UNIVERSITY OF KUOPIO, the Faculty of Natural and Environmental Sciences, Curriculum of Biochemistry USKI Juuso Oskari Thesis for Master of Science degree Supervisors Prof. Carsten Carlberg, Ph.D. Merja Heinäniemi, Ph.D. June 2008 Keywords: nuclear receptors; peroxisome proliferator-activated receptor; PPAR response element; apolipoprotein; lipid metabolism; high density lipoprotein; low density lipoprotein. Lipids are any fat-soluble, naturally-occurring molecules and one of their main biological functions is energy storage. Lipoproteins carry hydrophobic lipids in the water and salt-based blood environment for processing and energy supply in liver and other organs. In this study, the genomic area around the apolipoprotein genes was scanned in silico for PPAR response elements (PPREs) using the in vitro data-based computer program. Several new putative REs were found in surroundings of multiple lipoprotein genes. The responsiveness of those apolipoprotein genes to the PPAR ligands GW501516, rosiglitazone and GW7647 in the HepG2, HEK293 and THP-1 cell lines were tested with real-time PCR. The APOA1, APOA2, APOB, APOD, APOE, APOF, APOL1, APOL3, APOL5 and APOL6 genes were found to be regulated by PPARs in direct or secondary manners. Those results provide new insights in the understanding of lipid metabolism and so many lifestyle diseases like atherosclerosis, type 2 diabetes, heart disease and stroke. -

Mouse Net1 Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Net1 Knockout Project (CRISPR/Cas9) Objective: To create a Net1 knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Net1 gene (NCBI Reference Sequence: NM_019671 ; Ensembl: ENSMUSG00000021215 ) is located on Mouse chromosome 13. 12 exons are identified, with the ATG start codon in exon 1 and the TAA stop codon in exon 12 (Transcript: ENSMUST00000091853). Exon 4~11 will be selected as target site. Cas9 and gRNA will be co-injected into fertilized eggs for KO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Mice homozygous for a knock-out allele exhibit delayed mammary gland development during puberty associated with slower ductal extension, reduced ductal branching and epithelial cell proliferation, disorganized myoepithelial and ductal epithelial cells, and increased collagen deposition. Exon 4 starts from about 14.34% of the coding region. Exon 4~11 covers 63.25% of the coding region. The size of effective KO region: ~4176 bp. The KO region does not have any other known gene. Page 1 of 9 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele 5' gRNA region gRNA region 3' 1 4 5 6 7 8 9 10 11 12 Legends Exon of mouse Net1 Knockout region Page 2 of 9 https://www.alphaknockout.com Overview of the Dot Plot (up) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section upstream of Exon 4 is aligned with itself to determine if there are tandem repeats. Tandem repeats are found in the dot plot matrix. -

Chromosome 10, Frequently Lost in Human Melanoma, Encodes Multiple Tumor-Suppressive Functions
Published OnlineFirst January 22, 2014; DOI: 10.1158/0008-5472.CAN-13-1446 Cancer Tumor and Stem Cell Biology Research Chromosome 10, Frequently Lost in Human Melanoma, Encodes Multiple Tumor-Suppressive Functions Lawrence N. Kwong and Lynda Chin Abstract Although many DNA aberrations in melanoma have been well characterized, including focal amplification and deletions of oncogenes and tumor suppressors, broad regions of chromosomal gain and loss are less well understood. One possibility is that these broad events are a consequence of collateral damage from targeting single loci. Another possibility is that the loss of large regions permits the simultaneous repression of multiple tumor suppressors by broadly decreasing the resident gene dosage and expression. Here, we test this hypothesis in a targeted fashion using RNA interference to suppress multiple candidate residents in broad regions of loss. We find that loss of chromosome regions 6q, 10, and 11q21-ter is correlated with broadly decreased expression of most resident genes and that multiple resident genes impacted by broad regional loss of chromosome 10 are tumor suppressors capable of affecting tumor growth and/or invasion. We also provide additional functional support for Ablim1 as a novel tumor suppressor. Our results support the hypothesis that multiple cancer genes are targeted by regional chromosome copy number aberrations. Cancer Res; 74(6); 1814–21. Ó2014 AACR. Introduction mas (7). In contrast, the observed approximately 60% of CDKN2A/B Increasingly high-resolution genomic studies have estab- deletions on chromosome 9p are focal. These data PTEN lished that recurrent focal deletions and amplifications in suggest that although is a major driver of chromosome cancer can selectively target specific oncogenes and tumor 10 loss, other genes may also be targeted for inactivation. -

CD56+ T-Cells in Relation to Cytomegalovirus in Healthy Subjects and Kidney Transplant Patients
CD56+ T-cells in Relation to Cytomegalovirus in Healthy Subjects and Kidney Transplant Patients Institute of Infection and Global Health Department of Clinical Infection, Microbiology and Immunology Thesis submitted in accordance with the requirements of the University of Liverpool for the degree of Doctor in Philosophy by Mazen Mohammed Almehmadi December 2014 - 1 - Abstract Human T cells expressing CD56 are capable of tumour cell lysis following activation with interleukin-2 but their role in viral immunity has been less well studied. The work described in this thesis aimed to investigate CD56+ T-cells in relation to cytomegalovirus infection in healthy subjects and kidney transplant patients (KTPs). Proportions of CD56+ T cells were found to be highly significantly increased in healthy cytomegalovirus-seropositive (CMV+) compared to cytomegalovirus-seronegative (CMV-) subjects (8.38% ± 0.33 versus 3.29%± 0.33; P < 0.0001). In donor CMV-/recipient CMV- (D-/R-)- KTPs levels of CD56+ T cells were 1.9% ±0.35 versus 5.42% ±1.01 in D+/R- patients and 5.11% ±0.69 in R+ patients (P 0.0247 and < 0.0001 respectively). CD56+ T cells in both healthy CMV+ subjects and KTPs expressed markers of effector memory- RA T-cells (TEMRA) while in healthy CMV- subjects and D-/R- KTPs the phenotype was predominantly that of naïve T-cells. Other surface markers, CD8, CD4, CD58, CD57, CD94 and NKG2C were expressed by a significantly higher proportion of CD56+ T-cells in healthy CMV+ than CMV- subjects. Functional studies showed levels of pro-inflammatory cytokines IFN-γ and TNF-α, as well as granzyme B and CD107a were significantly higher in CD56+ T-cells from CMV+ than CMV- subjects following stimulation with CMV antigens. -

Statistical Methods for High-Dimensional Networked Data Analysis
Statistical Methods for High-Dimensional Networked Data Analysis by Yan Zhou A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Biostatistics) in The University of Michigan 2015 Doctoral Committee: Professor Peter X. K. Song, Chair Professor Matthias Kretzler Assistant Professor Xiaoquan William Wen Professor Ji Zhu c Yan Zhou 2015 All Rights Reserved To my family ii ACKNOWLEDGEMENTS I would like to acknowledge many people for their help throughout my graduate study. I would first like to express my deepest gratitude to my advisor Dr. Peter X.- K. Song, for sharing his knowledge, encouraging me to work hard and constantly try to improve my work, and giving me the freedom to explore a diverse set of projects. He is the one who first sparked my interest in networked data analysis. I am not only respectful for his immense knowledge, motivation, and enthusiasm towards research, but also appreciate his invaluable guidance and enthusiastic encouragement during many difficult moments in my research. I could not have imagined having a better advisor and mentor for my Ph.D. study. I would also like to thank my other thesis committee members Dr. Ji Zhu, Dr. Xiaoquan Willian Wen and Dr. Matthias Kretzler for their help and advices that made me more productive than what I could achieve on my own. All enlightening discussions I had with them have lead to better contents of this dissertation. In addition, I would like to acknowledge my collaborators, Dr. Pei Wang, Dr. Betsy Lozoff and Dr. Fengji Geng, who offered me excellent opportunities and con- structive suggestions to apply my statistical knowledge to solve real-world problems, which greatly strengthened the scientific background of my dissertation research. -
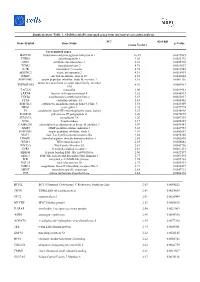
Supplementary Table 1. All Differentially Expressed Genes from Microarray Screening Analysis
Supplementary Table 1. All differentially expressed genes from microarray screening analysis. FCa (Gal-KD Gene Symbol Gene Name p-Value versus Vector) Up-regulated genes HAPLN1 hyaluronan and proteoglycan link protein 1 10.49 0.0027085 THBS1 thrombospondin 1 9.20 0.0022192 ODC1 ornithine decarboxylase 1 6.38 0.0055776 TGM2 transglutaminase 2 4.76 0.0015627 IL7R interleukin 7 receptor 4.75 0.0017245 SERINC2 serine incorporator 2 4.51 0.0014919 ITM2C integral membrane protein 2C 4.32 0.0044644 SERPINB7 serpin peptidase inhibitor, clade B, member 7 4.18 0.0081136 tumor necrosis factor receptor superfamily, member TNFRSF10D 4.01 0.0085561 10d TAGLN transgelin 3.90 0.0099963 LRRN4 leucine rich repeat neuronal 4 3.82 0.0046513 TGFB2 transforming growth factor beta 2 3.51 0.0035017 CPA4 carboxypeptidase A4 3.43 0.0008452 EPB41L3 erythrocyte membrane protein band 4.1-like 3 3.34 0.0025309 NRG1 neuregulin 1 3.28 0.0079724 F3 coagulation factor III (thromboplastin, tissue factor) 3.27 0.0038968 POLR3G polymerase III polypeptide G 3.26 0.0070675 SEMA7A semaphorin 7A 3.20 0.0087335 NT5E 5-nucleotidase 3.17 0.0036353 CAMK2N1 calmodulin-dependent protein kinase II inhibitor 1 3.07 0.0090141 TIMP3 TIMP metallopeptidase inhibitor 3 3.03 0.0047953 SERPINE1 serpin peptidase inhibitor, clade E 2.97 0.0053652 MALL mal, T-cell differentiation protein-like 2.88 0.0078205 DDAH1 dimethylarginine dimethylaminohydrolase 1 2.86 0.0002895 WDR3 WD repeat domain 3 2.85 0.0058842 WNT5A Wnt Family Member 5A 2.81 0.0043796 GPR1 G protein-coupled receptor 1 2.81 0.0021313 -

Abou Alezz, Monah Characterization of the Genomic and Splicing Features of Long 51 Non-Coding Rnas Using Bioinformatics Approaches
Posters A-Z Abou Alezz, Monah Characterization of the genomic and splicing features of long 51 non-coding RNAs using bioinformatics approaches Aguilera-Cortés, Paulina Identification of cellular lncRNAs associated to the HIV-1 genomic RNA 52 by interactome capture Ahram, Mamoun The androgen, dihydrotestosterone, induces chemoresistance of triple 53 negative breast cancer cells independent of ABCG2 and microRNA-328-3p Ahunbay, Esra Watching the self-catalyzed splicing of a group II intron with 54 single-molecule sensitivity Alemu, Endalkachew Integrated Global Mapping of miRNA:target-RNA Interactions 55 Ali, Tamer The lncRNA locus Handsdown regulates cardiac gene programs and is 56 essential for early mouse development Alzahrani, Salma Revisiting the sRNAome Under a Revised MicroRNA Annotation Criteria 57 Amorim, Marcella Dissecting the threshold-based readout of mobile small RNA gradients 58 Arnoldi, Michele SINEUPs technology: a new route to possibly treat 59 haploinsufficiency-induced Epilepsy and Autism Spectrum Disorders (ASDs) Azhikina, Tatyana Small RNA F6 influences Mycobacterium smegmatis entry into 60 dormancy EMBO | EMBL Symposium: The Non-Coding Genome Barasa, Sheila Olendo Long non-coding RNAs in High Grade Serous Ovarian Carcinoma 61 Behera, Alok LIN28 as a new drug target 62 Behrmann, Iris Modulation of the IL-6-Signaling Pathway in Liver Cells by miRNAs 63 Targeting gp130, JAK1, and/or STAT3 Bencurova, Petra Inhibition of miRNA-129-2-3p increases risk and severity of seizures in 64 developing brain Bica, Cecilia The emerging