Clinical Trial Protocol
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

ANTHRASIL™ Safely and Effectively
53 Weight-based Pediatric Dose Body Weight Vials per Dosea Body Weight Vials per Dose (kg) (kg) 1 HIGHLIGHTS OF PRESCRIBING INFORMATION <5 1 25 to <35 4 <10 1 35 to <50 5 10 to <18 2 50 to <60 6 These highlights do not include all the information needed to use 2 18 to <25 3 ≥60 7 ANTHRASIL™ safely and effectively. See full prescribing information for 3 aSelect initial dose based on clinical severity. Dose may be doubled for severe ANTHRASIL. 4 cases in patients >5 kg. 5 ANTHRASIL [Anthrax Immune Globulin Intravenous (Human)], sterile 54 6 Administer ANTHRASIL by slow intravenous infusion using an infusion solution for infusion 55 7 pump (maximum 2 mL per minute). 56 8 Initial U.S. Approval: March 24, 2015 57 9 ---------------------DOSAGE FORMS AND STRENGTHS---------------------- 10 58 59 Each single-use vial contains a minimum potency of ≥60 units by Toxin 11 WARNING: INTERACTIONS WITH GLUCOSE MONITORING 60 Neutralization Assay (TNA) (3). 12 SYSTEMS AND THROMBOSIS 61 13 See full prescribing information for complete boxed warning. 62 -------------------------------CONTRAINDICATIONS------------------------------ 14 • Maltose in immune globulin products, including ANTHRASIL, may give 63 • History of anaphylactic or severe systemic reaction to human immune 15 falsely high blood glucose levels with some blood point-of-care glucose 64 globulins (4) 16 testing systems (for example those based on the GDH-PQQ or glucose-dye- 65 • IgA deficiency with antibodies against IgA and a history of IgA 17 oxidoreductase methods) resulting in inappropriate administration of insulin 66 hypersensitivity (4) 18 and life-threatening hypoglycemia. To avoid interference by maltose 67 19 contained in ANTHRASIL, perform blood glucose measurements in patients 68 -----------------------WARNINGS AND PRECAUTIONS------------------------ 20 receiving ANTHRASIL with a glucose-specific method (monitor and test 21 strips). -

The Effect of Raxibacumab on the Immunogenicity of Anthrax Vaccine Adsorbed: a Phase
The effect of raxibacumab on the immunogenicity of anthrax vaccine adsorbed: a Phase IV, randomised, open-label, parallel-group, non-inferiority study Nancy Skoura, PhD1, Jie Wang-Jairaj, MD2, Oscar Della Pasqua MD2, Vijayalakshmi Chandrasekaran, MS1, Julia Billiard, PhD1, Anne Yeakey, MD3, William Smith, MD4, Helen Steel, MD2, Lionel K Tan, FRCP2 1GlaxoSmithKline, Inc. Collegeville, PA, USA 2GlaxoSmithKline. Stockley Park West, Middlesex, UK 3GlaxoSmithKline, Inc. Rockville, MD, USA 4AMR, at University of TN Medical Center, Knoxville, TN; New Orleans Center for Clinical Research (NOCCR), USA Author for Correspondence: Dr Lionel K Tan GlaxoSmithKline Stockley Park West 1–3 Ironbridge Road Uxbridge Middlesex UB11 1BT UK Email: [email protected] Tel: +44 (0)7341 079 683 1 Abstract: 340/350 Body text: 4321/4500 words including Research in Context Table/Figures: 2/4 References: 30 2 Abstract Background Raxibacumab is a monoclonal antibody (Ab) which binds protective antigen (PA) of Bacillus anthracis and is approved for treatment and post-exposure prophylaxis (PEP) of inhalational anthrax. Anthrax vaccine adsorbed (AVA), for anthrax prophylaxis, consists primarily of adsorbed PA. This post-approval study evaluated the effect of raxibacumab on immunogenicity of AVA. Methods In this open-label, parallel-group, non-inferiority study in three centres in the USA, healthy volunteers (aged 18–65 years) with no evidence of PA pre-exposure were randomised 1:1 to receive either subcutaneous 0·5 mL AVA on Days 1, 15, and 29 or raxibacumab intravenous infusion (40 mg/kg) immediately before AVA on Day 1, followed by AVA only on Days 15 and 29. -

A Review of the Efficacy of FDA-Approved B. Anthracis Anti
toxins Review A Review of the Efficacy of FDA-Approved B. anthracis Anti-Toxin Agents When Combined with Antibiotic or Hemodynamic Support in Infection- or Toxin-Challenged Preclinical Models Zoe Couse 1, Xizhong Cui 1, Yan Li 1, Mahtab Moayeri 2, Stephen Leppla 2 and Peter Q. Eichacker 1,* 1 Critical Care Medicine Department, Clinical Center, National Institutes of Health, Bethesda, MD 20892, USA; [email protected] (Z.C.); [email protected] (X.C.); [email protected] (Y.L.) 2 National Institutes of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD 20892, USA; [email protected] (M.M.); [email protected] (S.L.) * Correspondence: [email protected] Abstract: Anti-toxin agents for severe B. anthracis infection will only be effective if they add to the benefit of the two mainstays of septic shock management, antibiotic therapy and titrated hemody- namic support. Both of these standard therapies could negate benefits related to anti-toxin treatment. At present, three anthrax anti-toxin antibody preparations have received US Food and Drug Adminis- tration (FDA) approval: Raxibacumab, Anthrax Immune Globulin Intravenous (AIGIV) and ETI-204. Each agent is directed at the protective antigen component of lethal and edema toxin. All three agents were compared to placebo in antibiotic-treated animal models of live B. anthracis infection, and Raxibacumab and AIGIV were compared to placebo when combined with standard hemodynamic support in a 96 h canine model of anthrax toxin-associated shock. However, only AIG has actually Citation: Couse, Z.; Cui, X.; Li, Y.; been administered to a group of infected patients, and this experience was not controlled and offers Moayeri, M.; Leppla, S.; Eichacker, P.Q. -

The Project Bioshield Act: Issues for the 112Th Congress
The Project BioShield Act: Issues for the 112th Congress Frank Gottron Specialist in Science and Technology Policy March 13, 2012 Congressional Research Service 7-5700 www.crs.gov R42349 CRS Report for Congress Prepared for Members and Committees of Congress The Project BioShield Act: Issues for the 112th Congress Summary In 2004, Congress passed the Project BioShield Act (P.L. 108-276) to provide the federal government with new authorities related to the development, procurement, and use of medical countermeasures against chemical, biological, radiological, and nuclear (CBRN) terrorism agents. As the expiration of some of these authorities approaches, Congress is considering whether these authorities have sufficiently contributed to national preparedness to merit extension. The Project BioShield Act provides three main authorities: (1) guaranteeing a federal market for new CBRN medical countermeasures, (2) permitting emergency use of countermeasures that are either unapproved or have not been approved for the intended emergency use, and (3) relaxing regulatory requirements for some CBRN terrorism-related spending. The Department of Health and Human Services (HHS) has used each of these authorities. The HHS obligated approximately $2.5 billion to guarantee a government market for countermeasures against anthrax, botulism, radiation, and smallpox. The HHS allowed the emergency use of several unapproved products, including during the 2009 H1N1 influenza pandemic. The HHS used expedited review authorities to approve contracts and grants related to CBRN countermeasure research and development. The Department of Homeland Security (DHS) Appropriations Act, 2004 (P.L. 108-90) advance- appropriated $5.593 billion to acquire CBRN countermeasures through Project BioShield for FY2004-FY2013. Through FY2012, subsequent Congresses have removed $1.876 billion from this account through rescissions and transfers, more than one-third of the advance appropriation. -
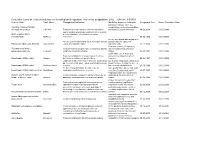
Designated Indication Cumulative List of All Products That Have Received
Cumulative List of all Products that have received Orphan Designation: Total active designations: 2002 Effecive: 5/5/2009 Generic Name Trade Name Designated Indication treatmentMarketing and/or Approved modification Indication of the Designated Date Market Exculsivity Date following conditions, which are Vaccinia Immune Globulin complications resulting from smallpox (Human) Intravenous CNJ-016 ForTreatment the control of complications and prevention of vacciniaof hemorrhagic vaccination episodes vaccination: Eczema vaccinatum 18.06.2004 01.05.9999 and for surgical prophylaxis in patients with hemophilia Antihemophilic factor A (congenital factor VIII deficiency or classic (recombinant) ReFacto hemophilia). 08.02.1996 01.01.9999 For use as a thyroid blocking agent in For use as a thyroid blocking agent in pediatric patients pediatric patients exposed to Potassium Iodide Oral Solution ThyroShield exposed to radiactive iodine radiactive iodine 17.11.2004 01.01.9999 Treatment (rescue) of respiratory Pulmonary surfactant For the treatment and prevention of respiratory distress distress syndrome in premature replacement, porcine Curosurf syndrome in premature infants. Longinfants. term treatment of children with 02.08.1993 01.01.9999 growth failure due to inadequate Treatment of idiopathic or organic growth hormone secretion of endogenous growth Somatropin (rDNA origin) Saizen deficiency in children with growth failure. Long-termhormone. treatment of growth failure 06.03.1987 01.01.9999 Long-term treatment of children who have growth failure due to a lack of adequate endogenous due to a lack of adequate endogenous growth hormone growth hormone secetion for once- or Somatropin (rDNA origin) Nutropin Depot secretion. twice-a-month administration. 28.10.1999 01.01.9999 Treatment of growth failure in children due to have growth failure due to inadequate Somatropin (rDNA origin) injection Norditropin inadequate growth hormone secretion. -

Ibm Micromedex® Carenotes Titles by Category
IBM MICROMEDEX® CARENOTES TITLES BY CATEGORY DECEMBER 2019 © Copyright IBM Corporation 2019 All company and product names mentioned are used for identification purposes only and may be trademarks of their respective owners. Table of Contents IBM Micromedex® CareNotes Titles by Category Allergy and Immunology ..................................................................................................................2 Ambulatory.......................................................................................................................................3 Bioterrorism ...................................................................................................................................18 Cardiology......................................................................................................................................18 Critical Care ...................................................................................................................................20 Dental Health .................................................................................................................................22 Dermatology ..................................................................................................................................23 Dietetics .........................................................................................................................................24 Endocrinology & Metabolic Disease ..............................................................................................26 -

The Project Bioshield Act: Issues for the 113Th Congress
The Project BioShield Act: Issues for the 113th Congress Frank Gottron Specialist in Science and Technology Policy June 18, 2014 Congressional Research Service 7-5700 www.crs.gov R43607 The Project BioShield Act: Issues for the 113th Congress Summary In 2004, Congress passed the Project BioShield Act (P.L. 108-276) to provide the federal government with new authorities related to the development, procurement, and use of medical countermeasures against chemical, biological, radiological, and nuclear (CBRN) terrorism agents. However, the government still lacks countermeasures against many of the CBRN terrorism agents determined by the government to pose the greatest threat. Congress is likely to consider whether modifications of these authorities or new authorities would help address remaining gaps. The authority generally referred to as Project BioShield allows the government to guarantee a market for CBRN medical countermeasures. Under this provision, the Secretary of Health and Human Services (HHS) may obligate funds to purchase countermeasures that still need up to 10 more years of development. Since 2004, HHS has obligated approximately $3.309 billion to guarantee a government market for countermeasures against anthrax, smallpox, botulism, radiation, and nerve agents. Another provision of the Project BioShield Act established a process through which the HHS Secretary may temporarily allow the emergency use of countermeasures that lack Food and Drug Administration (FDA) approval. The HHS has used this authority to allow the emergency use of unapproved products several times. The Department of Homeland Security (DHS) Appropriations Act, 2004 (P.L. 108-90) advance appropriated $5.593 billion to acquire CBRN countermeasures through Project BioShield between FY2004 and FY2013. -

DTT) Specification Guide
Data Translation Tool (DTT) Specification Guide 7.16.2020 This document provides specific instructions for setting up a file with the correct data elements (fields) for DTT. Table of Contents Permissions ......................................................................................................................................................................................... 4 File Format Elements ....................................................................................................................................................................... 4 Creating a Data File ........................................................................................................................................................................... 4 Import Profile Set Up ........................................................................................................................................................................................ 4 Patient Information ...................................................................................................................................................................................... 5 Patient and Vaccination Information .................................................................................................................................................... 6 Inventory Information ................................................................................................................................................................................ -
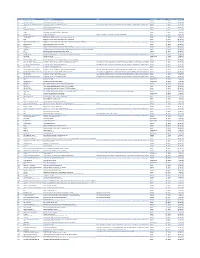
Cvx List.Pdf
CVX Code CVX Short Description Full Vaccine Name Note VaccineStatus internalID nonvaccine update_date 54 adenovirus, type 4 adenovirus vaccine, type 4, live, oral Inactive 1 False 28-May-10 55 adenovirus, type 7 adenovirus vaccine, type 7, live, oral Inactive 2 False 28-May-10 82 adenovirus, unspecified formulation adenovirus vaccine, unspecified formulation This CVX code allows reporting of a vaccination when formulation is unknown (for example, when reInactive 3 False 30-Sep-10 24 anthrax anthrax vaccine Active 4 False 11-Jun-19 19 BCG Bacillus Calmette-Guerin vaccine Active 5 False 28-May-10 27 botulinum antitoxin botulinum antitoxin Active 6 True 4-Sep-20 26 cholera, unspecified formulation cholera vaccine, unspecified formulation Inactive 7 False 17-Jun-16 29 CMVIG cytomegalovirus immune globulin, intravenous Active 8 True 4-Sep-20 56 dengue fever dengue fever vaccine Applies to dengue tetravalent product (e.g. DENGVAXIA) Active 9 False 24-Sep-19 12 diphtheria antitoxin diphtheria antitoxin Active 10 True 4-Sep-20 28 DT (pediatric) diphtheria and tetanus toxoids, adsorbed for pediatric use Active 11 False 28-May-10 20 DTaP diphtheria, tetanus toxoids and acellular pertussis vaccine Active 12 False 28-May-10 106 DTaP, 5 pertussis antigens diphtheria, tetanus toxoids and acellular pertussis vaccine, 5 pertussis antigens Active 13 False 28-May-10 107 DTaP, unspecified formulation diphtheria, tetanus toxoids and acellular pertussis vaccine, unspecified formulation This CVX code allows reporting of a vaccination when formulation is unknown -

Orphan Drug Designations and Approvals List As of 12-02-2013 Governs January 1, 2014
Orphan Drug Designations and Approvals List as of 12‐02‐2013 Governs January 1, 2014 ‐ March 31, 2014 Row Designation Generic Name Trade Name Orphan Designation Contact Company/Sponsor Num Date Treatment of Follicular Lymphoma, Small Lymphocytic Lymphoma, Lymphoplasmacytic Lymphoma, Splenic Marginal Zone Lymphoma, Extranodal Marginal Zone B‐cell Lymphoma of Mucosa‐ Associated Lymphoma Tissue (MALT), and Nodal Marginal Zone Lymphoma (Collectively Indolent B‐cell Non‐Hodgkin's 1 bendamustine hydrochloride Treanda 11/26/2013 Lymphoma) Cephalon, Inc. Treatment of subjects at risk of developing myelosuppression after a radiological or nuclear 2 Filgrastim Neupogen 11/20/2013 incident Amgen, Inc. adeno‐associated viral vector containing the human NADH Treatment of Leber Hereditary 3 Dehydrogenase 4 Gene n/a 11/20/2013 Optic Neuropathy Gen Sight Biologics Treatment of liver transplant recipients with reestablished fibrosis to delay the progression to cirrhosis and 4 emricasan n/a 11/20/2013 end stage liver disease Conatus Pharmaceuticals Inc. Page 1 of 262 Orphan Drug Designations and Approvals List as of 12‐02‐2013 Governs January 1, 2014 ‐ March 31, 2014 5‐((4‐bromo‐2‐ fluorophenyl)amino)‐4‐fluoro‐N‐(2‐ hydroxyethoxy)‐1‐methyl‐1H‐ Treatment Stage IIB‐IV Novartis Pharmaceuticals 5 benzo[d]imidazole‐6‐carboxamide n/a 11/19/2013 melanoma. Corporation Treatment of Hepatitis Delta 6 Lonafarnib n/a 11/19/2013 Virus (HDV)infection Eiger Biopharmaceuticals, Inc. Treatment in Stage IIB‐IV melanoma positive for the Novartis Pharmaceuticalues 7 MEK162 -

Monoclonal Antibody Therapies Against Anthrax
Toxins 2011, 3, 1004-1019; doi:10.3390/toxins3081004 OPEN ACCESS toxins ISSN 2072-6651 www.mdpi.com/journal/toxins Review Monoclonal Antibody Therapies against Anthrax Zhaochun Chen 1,*, Mahtab Moayeri 2 and Robert Purcell 1 1 Laboratory of Infectious Diseases National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD 20892, USA; E-Mail: [email protected] 2 Laboratory of Bacterial Diseases, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD 20892, USA; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-301-594-2308; Fax: +1-301-402-0524. Received: 10 June 2011; in revised form: 6 August 2011 / Accepted: 10 August 2011 / Published: 15 August 2011 Abstract: Anthrax is a highly lethal infectious disease caused by the spore-forming bacterium Bacillus anthracis. It not only causes natural infection in humans but also poses a great threat as an emerging bioterror agent. The lethality of anthrax is primarily attributed to the two major virulence factors: toxins and capsule. An extensive effort has been made to generate therapeutically useful monoclonal antibodies to each of the virulence components: protective antigen (PA), lethal factor (LF) and edema factor (EF), and the capsule of B. anthracis. This review summarizes the current status of anti-anthrax mAb development and argues for the potential therapeutic advantage of a cocktail of mAbs that recognize different epitopes or different virulence factors. Keywords: Bacillus anthracis; anti-PA mAbs; anti-LF mAbs; anti-EF mAbs; anti-capsule mAbs; post-exposure treatment of anthrax; a cocktail of mAbs 1. -

Anthrax Immune Globulin Improves Hemodynamics and Survival During B
Suffredini et al. Intensive Care Medicine Experimental (2017) 5:48 Intensive Care Medicine DOI 10.1186/s40635-017-0159-9 Experimental RESEARCH Open Access Anthrax immune globulin improves hemodynamics and survival during B. anthracis toxin-induced shock in canines receiving titrated fluid and vasopressor support Dante A. Suffredini1, Xizhong Cui1, Dharmvir Jaswal1, Kenneth E. Remy2, Yan Li1, Junfeng Sun1, Steven B. Solomon1, Yvonne Fitz1, Mahtab Moayeri3, Stephen Leppla3 and Peter Q. Eichacker1* * Correspondence: [email protected] Abstract 1Critical Care Medicine Department, Clinical Center, National Institutes of Background: Although anthrax immune globulin (AIG) improved survival in antibiotic- Health, Bldg 10, Rm 2C145, treated Bacillus anthracis-challenged animal models, whether it adds to the benefit of Bethesda, MD 20892, USA conventional hemodynamic support for B. anthracis toxin-associated shock is unknown. Full list of author information is available at the end of the article Methods: We therefore tested AIG in sedated, mechanically ventilated canines challenged with 24-h B. anthracis lethal and edema toxin infusions and supported for 96 h with a previously demonstrated protective regimen of titrated normal saline and norepinephrine. Results: Compared to controls, proportional survival (%) was increased with AIG treatment started 4 h before (33 vs. 100%, n = 6 each) or 2 h (17 vs. 86%, n =6and7respectively)or 5h(0vs.67%,n = 3 each) after the start of toxin (p ≤ 0.05) and overall [3 survivors of 15 controls (20%) vs. 14 of 16 AIG animals (88%); p = 0.006]. Averaged across treatment times, AIG increased blood pressure at 48 h and decreased norepinephrine requirements at 72 h (p ≤ 0.02), increased left ventricular ejection fraction at 48 and 72 h (p ≤ 0.02), and increased urine output and decreased net fluid balance at 72 and 96 h (p ≤ 0.04).