The Effect of Raxibacumab on the Immunogenicity of Anthrax Vaccine Adsorbed: a Phase
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

AHFS Pharmacologic-Therapeutic Classification System
AHFS Pharmacologic-Therapeutic Classification System Abacavir 48:24 - Mucolytic Agents - 382638 8:18.08.20 - HIV Nucleoside and Nucleotide Reverse Acitretin 84:92 - Skin and Mucous Membrane Agents, Abaloparatide 68:24.08 - Parathyroid Agents - 317036 Aclidinium Abatacept 12:08.08 - Antimuscarinics/Antispasmodics - 313022 92:36 - Disease-modifying Antirheumatic Drugs - Acrivastine 92:20 - Immunomodulatory Agents - 306003 4:08 - Second Generation Antihistamines - 394040 Abciximab 48:04.08 - Second Generation Antihistamines - 394040 20:12.18 - Platelet-aggregation Inhibitors - 395014 Acyclovir Abemaciclib 8:18.32 - Nucleosides and Nucleotides - 381045 10:00 - Antineoplastic Agents - 317058 84:04.06 - Antivirals - 381036 Abiraterone Adalimumab; -adaz 10:00 - Antineoplastic Agents - 311027 92:36 - Disease-modifying Antirheumatic Drugs - AbobotulinumtoxinA 56:92 - GI Drugs, Miscellaneous - 302046 92:20 - Immunomodulatory Agents - 302046 92:92 - Other Miscellaneous Therapeutic Agents - 12:20.92 - Skeletal Muscle Relaxants, Miscellaneous - Adapalene 84:92 - Skin and Mucous Membrane Agents, Acalabrutinib 10:00 - Antineoplastic Agents - 317059 Adefovir Acamprosate 8:18.32 - Nucleosides and Nucleotides - 302036 28:92 - Central Nervous System Agents, Adenosine 24:04.04.24 - Class IV Antiarrhythmics - 304010 Acarbose Adenovirus Vaccine Live Oral 68:20.02 - alpha-Glucosidase Inhibitors - 396015 80:12 - Vaccines - 315016 Acebutolol Ado-Trastuzumab 24:24 - beta-Adrenergic Blocking Agents - 387003 10:00 - Antineoplastic Agents - 313041 12:16.08.08 - Selective -

ANTHRASIL™ Safely and Effectively
53 Weight-based Pediatric Dose Body Weight Vials per Dosea Body Weight Vials per Dose (kg) (kg) 1 HIGHLIGHTS OF PRESCRIBING INFORMATION <5 1 25 to <35 4 <10 1 35 to <50 5 10 to <18 2 50 to <60 6 These highlights do not include all the information needed to use 2 18 to <25 3 ≥60 7 ANTHRASIL™ safely and effectively. See full prescribing information for 3 aSelect initial dose based on clinical severity. Dose may be doubled for severe ANTHRASIL. 4 cases in patients >5 kg. 5 ANTHRASIL [Anthrax Immune Globulin Intravenous (Human)], sterile 54 6 Administer ANTHRASIL by slow intravenous infusion using an infusion solution for infusion 55 7 pump (maximum 2 mL per minute). 56 8 Initial U.S. Approval: March 24, 2015 57 9 ---------------------DOSAGE FORMS AND STRENGTHS---------------------- 10 58 59 Each single-use vial contains a minimum potency of ≥60 units by Toxin 11 WARNING: INTERACTIONS WITH GLUCOSE MONITORING 60 Neutralization Assay (TNA) (3). 12 SYSTEMS AND THROMBOSIS 61 13 See full prescribing information for complete boxed warning. 62 -------------------------------CONTRAINDICATIONS------------------------------ 14 • Maltose in immune globulin products, including ANTHRASIL, may give 63 • History of anaphylactic or severe systemic reaction to human immune 15 falsely high blood glucose levels with some blood point-of-care glucose 64 globulins (4) 16 testing systems (for example those based on the GDH-PQQ or glucose-dye- 65 • IgA deficiency with antibodies against IgA and a history of IgA 17 oxidoreductase methods) resulting in inappropriate administration of insulin 66 hypersensitivity (4) 18 and life-threatening hypoglycemia. To avoid interference by maltose 67 19 contained in ANTHRASIL, perform blood glucose measurements in patients 68 -----------------------WARNINGS AND PRECAUTIONS------------------------ 20 receiving ANTHRASIL with a glucose-specific method (monitor and test 21 strips). -

Phage Display-Based Strategies for Cloning and Optimization of Monoclonal Antibodies Directed Against Human Pathogens
Int. J. Mol. Sci. 2012, 13, 8273-8292; doi:10.3390/ijms13078273 OPEN ACCESS International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms Review Phage Display-based Strategies for Cloning and Optimization of Monoclonal Antibodies Directed against Human Pathogens Nicola Clementi *,†, Nicasio Mancini †, Laura Solforosi, Matteo Castelli, Massimo Clementi and Roberto Burioni Microbiology and Virology Unit, “Vita-Salute” San Raffaele University, Milan 20132, Italy; E-Mails: [email protected] (N.M.); [email protected] (L.S.); [email protected] (M.C.); [email protected] (M.C.); [email protected] (R.B.) † These authors contributed equally to this work. * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +39-2-2643-5082; Fax: +39-2-2643-4288. Received: 16 March 2012; in revised form: 25 June 2012 / Accepted: 27 June 2012 / Published: 4 July 2012 Abstract: In the last two decades, several phage display-selected monoclonal antibodies (mAbs) have been described in the literature and a few of them have managed to reach the clinics. Among these, the anti-respiratory syncytial virus (RSV) Palivizumab, a phage-display optimized mAb, is the only marketed mAb directed against microbial pathogens. Palivizumab is a clear example of the importance of choosing the most appropriate strategy when selecting or optimizing an anti-infectious mAb. From this perspective, the extreme versatility of phage-display technology makes it a useful tool when setting up different strategies for the selection of mAbs directed against human pathogens, especially when their possible clinical use is considered. -

Version 12.0, 01 Jun 2018 ANNEX I SUMMARY of PRODUCT
BioThrax® (Anthrax Vaccine Adsorbed) 1.3.1 Summary of Product Characteristics, Labelling and Package Leaflet Version 12.0, 01 Jun 2018 ANNEX I SUMMARY OF PRODUCT CHARACTERISTICS, LABELLING AND PACKAGE LEAFLET Emergent BioSolutions, Inc. Confidential and Proprietary Page 1 of 25 BioThrax® (Anthrax Vaccine Adsorbed) 1.3.1 Summary of Product Characteristics, Labelling and Package Leaflet SUMMARY OF PRODUCT CHARACTERISTICS Emergent BioSolutions, Inc. Confidential and Proprietary Page 2 of 25 BioThrax® (Anthrax Vaccine Adsorbed) 1.3.1 Summary of Product Characteristics, Labelling and Package Leaflet This medicinal product is subject to additional monitoring. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions. See Section 4.8 for how to report adverse reactions 1 NAME OF THE MEDICINAL PRODUCT BioThrax1 suspension for injection. Anthrax Vaccine Adsorbed (purified cell-free filtrate) 2 QUALITATIVE AND QUANTITATIVE COMPOSITION One dose (0.5 mL) contains: Anthrax antigen filtrate: 50 micrograms (50 mcg) a, b For a full list of excipients, see Section 6.1. a Produced from cell-free filtrates of an avirulent strain of Bacillus anthracis b Adsorbed on aluminium hydroxide, hydrated (0.6 mg Al3+) 3 PHARMACEUTICAL FORM Suspension for injection. Sterile, milky-white liquid suspension, when mixed. 4 CLINICAL PARTICULARS 4.1 Therapeutic indications BioThrax is indicated for the prevention of disease caused by Bacillus anthracis, in adults at risk of exposure. BioThrax should be used in accordance with official recommendations, where available. 4.2 Posology and method of administration Posology: Primary Immunisation: 3-doses each of 0.5 mL, given at 0, 1 and 6 months. -

A Review of the Efficacy of FDA-Approved B. Anthracis Anti
toxins Review A Review of the Efficacy of FDA-Approved B. anthracis Anti-Toxin Agents When Combined with Antibiotic or Hemodynamic Support in Infection- or Toxin-Challenged Preclinical Models Zoe Couse 1, Xizhong Cui 1, Yan Li 1, Mahtab Moayeri 2, Stephen Leppla 2 and Peter Q. Eichacker 1,* 1 Critical Care Medicine Department, Clinical Center, National Institutes of Health, Bethesda, MD 20892, USA; [email protected] (Z.C.); [email protected] (X.C.); [email protected] (Y.L.) 2 National Institutes of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD 20892, USA; [email protected] (M.M.); [email protected] (S.L.) * Correspondence: [email protected] Abstract: Anti-toxin agents for severe B. anthracis infection will only be effective if they add to the benefit of the two mainstays of septic shock management, antibiotic therapy and titrated hemody- namic support. Both of these standard therapies could negate benefits related to anti-toxin treatment. At present, three anthrax anti-toxin antibody preparations have received US Food and Drug Adminis- tration (FDA) approval: Raxibacumab, Anthrax Immune Globulin Intravenous (AIGIV) and ETI-204. Each agent is directed at the protective antigen component of lethal and edema toxin. All three agents were compared to placebo in antibiotic-treated animal models of live B. anthracis infection, and Raxibacumab and AIGIV were compared to placebo when combined with standard hemodynamic support in a 96 h canine model of anthrax toxin-associated shock. However, only AIG has actually Citation: Couse, Z.; Cui, X.; Li, Y.; been administered to a group of infected patients, and this experience was not controlled and offers Moayeri, M.; Leppla, S.; Eichacker, P.Q. -

The Project Bioshield Act: Issues for the 112Th Congress
The Project BioShield Act: Issues for the 112th Congress Frank Gottron Specialist in Science and Technology Policy March 13, 2012 Congressional Research Service 7-5700 www.crs.gov R42349 CRS Report for Congress Prepared for Members and Committees of Congress The Project BioShield Act: Issues for the 112th Congress Summary In 2004, Congress passed the Project BioShield Act (P.L. 108-276) to provide the federal government with new authorities related to the development, procurement, and use of medical countermeasures against chemical, biological, radiological, and nuclear (CBRN) terrorism agents. As the expiration of some of these authorities approaches, Congress is considering whether these authorities have sufficiently contributed to national preparedness to merit extension. The Project BioShield Act provides three main authorities: (1) guaranteeing a federal market for new CBRN medical countermeasures, (2) permitting emergency use of countermeasures that are either unapproved or have not been approved for the intended emergency use, and (3) relaxing regulatory requirements for some CBRN terrorism-related spending. The Department of Health and Human Services (HHS) has used each of these authorities. The HHS obligated approximately $2.5 billion to guarantee a government market for countermeasures against anthrax, botulism, radiation, and smallpox. The HHS allowed the emergency use of several unapproved products, including during the 2009 H1N1 influenza pandemic. The HHS used expedited review authorities to approve contracts and grants related to CBRN countermeasure research and development. The Department of Homeland Security (DHS) Appropriations Act, 2004 (P.L. 108-90) advance- appropriated $5.593 billion to acquire CBRN countermeasures through Project BioShield for FY2004-FY2013. Through FY2012, subsequent Congresses have removed $1.876 billion from this account through rescissions and transfers, more than one-third of the advance appropriation. -

Whether Section 564 of the Food, Drug, and Cosmetic Act Prohibits Entities from Requiring the Use of a Vaccine Subject to an Emergency Use Authorization
(Slip Opinion) Whether Section 564 of the Food, Drug, and Cosmetic Act Prohibits Entities from Requiring the Use of a Vaccine Subject to an Emergency Use Authorization Section 564(e)(1)(A)(ii)(III) of the Food, Drug, and Cosmetic Act concerns only the provision of information to potential vaccine recipients and does not prohibit public or private entities from imposing vaccination requirements for a vaccine that is subject to an emergency use authorization. July 6, 2021 MEMORANDUM OPINION FOR THE DEPUTY COUNSEL TO THE PRESIDENT Section 564 of the Food, Drug, and Cosmetic Act (“FDCA”), 21 U.S.C. § 360bbb-3,1 authorizes the Food and Drug Administration (“FDA”) to issue an “emergency use authorization” (“EUA”) for a medical product, such as a vaccine, under certain emergency circumstances. This authoriza- tion permits the product to be introduced into interstate commerce and administered to individuals even when FDA has not approved the product for more general distribution pursuant to its standard review process. Section 564 directs FDA—“to the extent practicable” given the emergen- cy circumstances and “as the [agency] finds necessary or appropriate to protect the public health”—to impose “[a]ppropriate” conditions on each EUA. FDCA § 564(e)(1)(A). Some of these conditions are designed to ensure that recipients of the product “are informed” of certain things, including “the option to accept or refuse administration of the product.” Id. § 564(e)(1)(A)(ii)(III). Since December 2020, FDA has granted EUAs for three vaccines to prevent coronavirus disease 2019 (“COVID-19”). In each of these author- izations, FDA imposed the “option to accept or refuse” condition by requiring the distribution to potential vaccine recipients of a Fact Sheet that states: “It is your choice to receive or not receive [the vaccine]. -

RAXIBACUMAB Safely and Effectively
HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all of the information needed to use RAXIBACUMAB safely and effectively. See full prescribing information --------------------DOSAGE FORMS AND STRENGTHS---------------- for RAXIBACUMAB. Single-use vial contains 1700 mg/34 mL (50 mg/mL) raxibacumab solution. (3) RAXIBACUMAB injection, for intravenous use Initial U.S. Approval: 2012 ----------------------------- CONTRAINDICATIONS--- ---------------------- None. (4) -------------------------- INDICATIONS AND USAGE--- ------------------- Raxibacumab is indicated for the treatment of adult and pediatric patients with ----------------------WARNINGS AND PRECAUTIONS--- --------------- inhalational anthrax due to Bacillus anthracis in combination with appropriate Infusion reactions may occur. Premedicate with diphenhydramine. Slow or antibacterial drugs, and for prophylaxis of inhalational anthrax when interrupt infusion and administer treatment based on severity of the reaction. alternative therapies are not available or are not appropriate. (1) (5.1) Limitations of Use: The effectiveness of raxibacumab is based solely on efficacy studies in -----------------------------ADVERSE REACTIONS--- ---------------------- animal models of inhalational anthrax. (1.2, 14.1) Common adverse reactions in healthy adult subjects (≥1.5%) were: rash, pain There have been no studies of raxibacumab in the pediatric population. in extremity, pruritus, and somnolence. (6.1) Dosing in pediatric patients was derived using a population PK approach. -

Studies on Antitoxin Use for Postexposure Prophylaxis (PEP)
ACIP Anthrax Vaccine Work Group William Bower, MD, FIDSA Epidemiology Team Lead Bacterial Special Pathogens October 23, 2018 National Center for Emerging and Zoonotic Disease Division of High-Consequence Pathogens and Pathology Outline Overview of anthrax antitoxins Studies on antitoxin use for postexposure prophylaxis (PEP) . Anthrax antitoxin for PEP and survival in animal models . Anthrax antitoxin effect on acquired immunity . Anthrax antitoxin use with anthrax vaccine adsorbed (AVA) Work group discussions Guidance on anthrax antitoxin use for PEP 2 Anthrax Pathogenesis 3 Background – Anthrax Antitoxins There are currently three FDA approved anthrax antitoxins RAXIBACUMAB . Initial U.S. Approval: 2012 . Human IgG1λ monoclonal antibody that binds the protective antigen (PA) component of Bacillus anthracis toxin ANTHRASIL (Anthrax Immune Globulin Intravenous (AIGIV)) . Initial U.S. Approval: 2015 . Purified human IgG containing polyclonal antibodies that bind the protective antigen (PA) component of B. anthracis lethal and edema toxins collected from individuals immunized with AVA ANTHIM (obiltoxaximab) . Initial U.S. Approval: 2016 . Chimeric IgG1 kappa monoclonal antibody that binds the PA component of B. anthracis toxin 4 Background – Anthrax Antitoxins Indications Anthrax antitoxins all have an indication for the treatment of adult and pediatric patients with inhalation anthrax due to B. anthracis in combination with appropriate antimicrobials The monoclonal antitoxins, raxibacumab and obiltoxaximab, are also indicated for -

Boosting Anti-Infective Antibody and Vaccine Development
ADVERTISEMENT FEATURE YUMAB GmbH www.yumab.com YUMAB: boosting anti-infective antibody and vaccine development YUMAB, a global provider of fully human monoclonal antibody discovery and development, has relocated its headquarters to Germany’s hotspot of infectious research and is advancing new opportunities for the expedited development of prophylactic and therapeutic antibodies and vaccines to fight infectious diseases. Biotechnology company YUMAB, a global provider The combination of deep expertise in infectious of fully human monoclonal antibody (mAb) dis- Therapeutics disease and an advanced fully human mAb discov- covery and development, has recently relocated its ery and development platform makes YUMAB an profile headquarters and research and development labs to ideal partner to drive infectious disease programs Science Campus Braunschweig-South, Germany’s Pathogens YUMAB® PLATFORM Vaccines from basic research to clinical translation. The fully infectious disease research hotspot. YUMAB is now human mAbs developed at YUMAB can be tailored colocated with the Helmholtz Centre of Infection to the specific needs of any kind of mAb develop- Diagnostics Research (HZI), the Leibniz Institute-German ment program, at any stage, and with the properties Collection of Microorganisms and Cell Cultures needed for novel prophylactic or therapeutic strate- (DSMZ) and the German Centre for Infection Research gies against infectious diseases. (DZIF): three world leaders in the fight against infec- Fig. 1| Driving anti-infective mAbs with YUMAB’s tious diseases that provide an ideal environment for fully human mAb and target discovery and Partnering to fight infectious diseases YUMAB to expand its global collaboration network development platform. YUMAB is committed to accelerating the develop- in this space. -
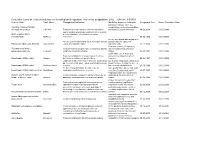
Designated Indication Cumulative List of All Products That Have Received
Cumulative List of all Products that have received Orphan Designation: Total active designations: 2002 Effecive: 5/5/2009 Generic Name Trade Name Designated Indication treatmentMarketing and/or Approved modification Indication of the Designated Date Market Exculsivity Date following conditions, which are Vaccinia Immune Globulin complications resulting from smallpox (Human) Intravenous CNJ-016 ForTreatment the control of complications and prevention of vacciniaof hemorrhagic vaccination episodes vaccination: Eczema vaccinatum 18.06.2004 01.05.9999 and for surgical prophylaxis in patients with hemophilia Antihemophilic factor A (congenital factor VIII deficiency or classic (recombinant) ReFacto hemophilia). 08.02.1996 01.01.9999 For use as a thyroid blocking agent in For use as a thyroid blocking agent in pediatric patients pediatric patients exposed to Potassium Iodide Oral Solution ThyroShield exposed to radiactive iodine radiactive iodine 17.11.2004 01.01.9999 Treatment (rescue) of respiratory Pulmonary surfactant For the treatment and prevention of respiratory distress distress syndrome in premature replacement, porcine Curosurf syndrome in premature infants. Longinfants. term treatment of children with 02.08.1993 01.01.9999 growth failure due to inadequate Treatment of idiopathic or organic growth hormone secretion of endogenous growth Somatropin (rDNA origin) Saizen deficiency in children with growth failure. Long-termhormone. treatment of growth failure 06.03.1987 01.01.9999 Long-term treatment of children who have growth failure due to a lack of adequate endogenous due to a lack of adequate endogenous growth hormone growth hormone secetion for once- or Somatropin (rDNA origin) Nutropin Depot secretion. twice-a-month administration. 28.10.1999 01.01.9999 Treatment of growth failure in children due to have growth failure due to inadequate Somatropin (rDNA origin) injection Norditropin inadequate growth hormone secretion. -

Considerations for Anthrax Vaccine Adsorbed (Ava) Post- Exposure Prioritization
2013 The Centers for Disease Control and Prevention FINAL CONSIDERATIONS FOR ANTHRAX VACCINE ADSORBED (AVA) POST- EXPOSURE PRIORITIZATION FINAL DISCLAIMER: This document considers prioritization of Anthrax Vaccine Adsorbed (AVA) in a post-event setting. This document does not include implementation considerations for post-event vaccination. This document does not include policy or implementation considerations for pre-event vaccination. This document does not include policy considerations for reserving vaccine in a post-event setting (e.g., holding back vaccine because of high risk of subsequent event). This document does not address vaccination for long-term exposure risks to Bacillus anthracis spores (> 6 months). The principles in this document apply regardless of the number of doses available. TABLE OF CONTENTS PRELUDE ...................................................................................................................................................... 1 1. BACKGROUND ON ANTHRAX AND AVA ................................................................................................ 2 1.1 Information about Anthrax Vaccine Adsorbed (AVA) .................................................................. 2 1.2 Anthrax as a Public Health Concern ............................................................................................ 3 2. AVA PRIORITIZATION GUIDANCE BACKGROUND AND METHODS ........................................................... 4 3. DEFINITIONS AND MAJOR CONCEPTS..................................................................................................