Whether Section 564 of the Food, Drug, and Cosmetic Act Prohibits Entities from Requiring the Use of a Vaccine Subject to an Emergency Use Authorization
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Logical Fallacies Moorpark College Writing Center
Logical Fallacies Moorpark College Writing Center Ad hominem (Argument to the person): Attacking the person making the argument rather than the argument itself. We would take her position on child abuse more seriously if she weren’t so rude to the press. Ad populum appeal (appeal to the public): Draws on whatever people value such as nationality, religion, family. A vote for Joe Smith is a vote for the flag. Alleged certainty: Presents something as certain that is open to debate. Everyone knows that… Obviously, It is obvious that… Clearly, It is common knowledge that… Certainly, Ambiguity and equivocation: Statements that can be interpreted in more than one way. Q: Is she doing a good job? A: She is performing as expected. Appeal to fear: Uses scare tactics instead of legitimate evidence. Anyone who stages a protest against the government must be a terrorist; therefore, we must outlaw protests. Appeal to ignorance: Tries to make an incorrect argument based on the claim never having been proven false. Because no one has proven that food X does not cause cancer, we can assume that it is safe. Appeal to pity: Attempts to arouse sympathy rather than persuade with substantial evidence. He embezzled a million dollars, but his wife had just died and his child needed surgery. Begging the question/Circular Logic: Proof simply offers another version of the question itself. Wrestling is dangerous because it is unsafe. Card stacking: Ignores evidence from the one side while mounting evidence in favor of the other side. Users of hearty glue say that it works great! (What is missing: How many users? Great compared to what?) I should be allowed to go to the party because I did my math homework, I have a ride there and back, and it’s at my friend Jim’s house. -

United States Code
United States Code COMPREHENSIVE COVERAGE DATING BACK TO INCEPTION IN 1925-1926, IN A FULLY SEARCHABLE, USER-FRIENDLY FORMAT! The United States Code is a consolidation and codification by subject matter of the general and permanent laws of the United States. The Office of the Law Revision Counsel of the U.S. House of Representatives prepares and publishes the United States Code pursuant to section 285b of title 2 of the Code. The Code does not include regulations issues by executive branch agencies, decisions of the Federal courts, treaties, or laws enacted by State or local governments. THE HEINONLINE ADVANTAGE HeinOnline’s version of the United States Code provides users with a single source for the entire archive along with current content of the United States Code. • Comprehensive coverage from inception • Browse by Title or Edition • Quickly find a document with the custom citation locator • Content is easy to both browse and search • Powerful search engine enables users to locate topic-specific content quickly and easily • As part of several Core packages, the U.S. Code in HeinOnline provides an incredible platform for an even better value! United States Code INCLUDES EARLY FEDERAL CODES & COMPILATIONS OF STATUTES The Early Federal Laws Collection represents the most complete collection of federal statute compilations, prior to the United States Code in 1926. The collection includes the first compilation by Richard Folwell (1795- 1814), the Bioren and Duane editions (1815), Thomas Herty Digest (1800), William Graydon’s Abridgement (1803), among others. Browse the laws by Title, Coverage, Publication Date, Volumes, Congress, or Compiler, using a custom chart of documents. -

Hallucinogens - LSD, Peyote, Psilocybin, and PCP
Information for Behavioral Health Providers in Primary Care Hallucinogens - LSD, Peyote, Psilocybin, and PCP What are Hallucinogens? Hallucinogenic compounds found in some plants and mushrooms (or their extracts) have been used— mostly during religious rituals—for centuries. Almost all hallucinogens contain nitrogen and are classified as alkaloids. Many hallucinogens have chemical structures similar to those of natural neurotransmitters (e.g., acetylcholine-, serotonin-, or catecholamine-like). While the exact mechanisms by which hallucinogens exert their effects remain unclear, research suggests that these drugs work, at least partially, by temporarily interfering with neurotransmitter action or by binding to their receptor sites. This InfoFacts will discuss four common types of hallucinogens: LSD (d-lysergic acid diethylamide) is one of the most potent mood-changing chemicals. It was discovered in 1938 and is manufactured from lysergic acid, which is found in ergot, a fungus that grows on rye and other grains. Peyote is a small, spineless cactus in which the principal active ingredient is mescaline. This plant has been used by natives in northern Mexico and the southwestern United States as a part of religious ceremonies. Mescaline can also be produced through chemical synthesis. Psilocybin (4-phosphoryloxy-N, N-dimethyltryptamine) is obtained from certain types of mushrooms that are indigenous to tropical and subtropical regions of South America, Mexico, and the United States. These mushrooms typically contain less than 0.5 percent psilocybin plus trace amounts of psilocin, another hallucinogenic substance. PCP (phencyclidine) was developed in the 1950s as an intravenous anesthetic. Its use has since been discontinued due to serious adverse effects. How Are Hallucinogens Abused? The very same characteristics that led to the incorporation of hallucinogens into ritualistic or spiritual traditions have also led to their propagation as drugs of abuse. -

Drug and Alcohol Abuse Prevention Handbook FOREWARD
Drug and Alcohol Abuse Prevention Handbook FOREWARD Grayson College recognizes that the illicit use of drugs and/or the abuse of alcohol are a persistent health problem of major proportion affecting our society physically, mentally, and socially. Illicit drug use and /or alcohol abuse can adversely affect an individual’s personal life, safety, health, and mental and physical performance. It is the intent of GC to provide employees and students pertinent information related to illicit drug use and/or alcohol abuse in an effort to prevent such harm. GC is committed to promoting and maintaining a work and academic environment that is free from illegal alcohol and drug use and abuse, in accordance with all federal, state, and local laws. Students, employees, and visitors are prohibited from possessing, consuming, manufacturing, dispensing, or being under the influence of alcohol/illegal drugs or engaging in improper self- medication while on college property or college business. Any member of the college community who violates this policy is subject to both prosecution and punishment under federal, state, and local laws to disciplinary proceedings by the college. This alcohol/drug policy is not designed to punish people for seeking rehabilitation. All information about those individuals who voluntarily avail themselves of drug or alcohol counseling or rehabilitation will not be used as a basis for disciplinary action or be used against an individual in any way. College employees and students who violate the alcohol/drug policy shall be informed about and referred to services to assist them in determining whether they are abusing drugs and alcohol or are chemically dependent. -

Bias and Critical Thinking
BIAS AND CRITICAL THINKING Point: there is an alternative to • being “biased” (one-sided, closed-minded, etc.) • simply having an “opinion” (by which I mean a viewpoint that has subjective value only: “everyone has their own opinions”) • being neutral and not taking a position In thinking about bias, it is important to distinguish between four things: 1. a particular position taken on an issue 2. the source of that position (its support and basis) 3. the resistance or openness to other positions 4. the impact that position has on other positions and viewpoints taken by the person Too often, people confuse these four. One result is that people sometimes assume that taking any position on an issue (#1) is an indication of bias. If this were true, then the only way to avoid bias would be to not take a position but rather simply present what are considered to be facts. In this way one is supposedly “objective” and “neutral.” However, it is highly debatable whether one can really be objective and neutral or whether one can present objective facts in a completely neutral way. More importantly, there are two troublesome implications of such a viewpoint on bias: • the ideal would seem to be not taking a position (but to really deal with issues we have to take a position) • all positions are biased and therefore it is difficult if not impossible to judge one position superior to another. It is far better to reject the idea that taking any position always implies bias. Rather, bias is a function either of the source of that position, or the resistance one has to other positions, or the impact that position has on other positions and viewpoints taken. -

Hallucinogens and Dissociative Drugs
Long-Term Effects of Hallucinogens See page 5. from the director: Research Report Series Hallucinogens and dissociative drugs — which have street names like acid, angel dust, and vitamin K — distort the way a user perceives time, motion, colors, sounds, and self. These drugs can disrupt a person’s ability to think and communicate rationally, or even to recognize reality, sometimes resulting in bizarre or dangerous behavior. Hallucinogens such as LSD, psilocybin, peyote, DMT, and ayahuasca cause HALLUCINOGENS AND emotions to swing wildly and real-world sensations to appear unreal, sometimes frightening. Dissociative drugs like PCP, DISSOCIATIVE DRUGS ketamine, dextromethorphan, and Salvia divinorum may make a user feel out of Including LSD, Psilocybin, Peyote, DMT, Ayahuasca, control and disconnected from their body PCP, Ketamine, Dextromethorphan, and Salvia and environment. In addition to their short-term effects What Are on perception and mood, hallucinogenic Hallucinogens and drugs are associated with psychotic- like episodes that can occur long after Dissociative Drugs? a person has taken the drug, and dissociative drugs can cause respiratory allucinogens are a class of drugs that cause hallucinations—profound distortions depression, heart rate abnormalities, and in a person’s perceptions of reality. Hallucinogens can be found in some plants and a withdrawal syndrome. The good news is mushrooms (or their extracts) or can be man-made, and they are commonly divided that use of hallucinogenic and dissociative Hinto two broad categories: classic hallucinogens (such as LSD) and dissociative drugs (such drugs among U.S. high school students, as PCP). When under the influence of either type of drug, people often report rapid, intense in general, has remained relatively low in emotional swings and seeing images, hearing sounds, and feeling sensations that seem real recent years. -

Form I-589, Application for Asylum and for Withholding of Removal
Department of Homeland Security OMB No. 1615-0067; Expires 07/31/2022 U.S. Citizenship and Immigration Services U.S. Department of Justice I-589, Application for Asylum Executive Office for Immigration Review and for Withholding of Removal START HERE - Type or print in black ink. See the instructions for information about eligibility and how to complete and file this application. There is no filing fee for this application. NOTE: Check this box if you also want to apply for withholding of removal under the Convention Against Torture. Part A.I. Information About You 1. Alien Registration Number(s) (A-Number) (if any) 2. U.S. Social Security Number (if any) 3. USCIS Online Account Number (if any) 4. Complete Last Name 5. First Name 6. Middle Name 7. What other names have you used (include maiden name and aliases)? 8. Residence in the U.S. (where you physically reside) Street Number and Name Apt. Number City State Zip Code Telephone Number ( ) 9. Mailing Address in the U.S. (if different than the address in Item Number 8) In Care Of (if applicable): Telephone Number ( ) Street Number and Name Apt. Number City State Zip Code 10. Gender: Male Female 11. Marital Status: Single Married Divorced Widowed 12. Date of Birth (mm/dd/yyyy) 13. City and Country of Birth 14. Present Nationality (Citizenship) 15. Nationality at Birth 16. Race, Ethnic, or Tribal Group 17. Religion 18. Check the box, a through c, that applies: a. I have never been in Immigration Court proceedings. b. I am now in Immigration Court proceedings. -

Drugs That Can Cause Delirium (Anticholinergic / Toxic Metabolites)
Drugs that can Cause Delirium (anticholinergic / toxic metabolites) Deliriants (drugs causing delirium) Prescription drugs . Central acting agents – Sedative hypnotics (e.g., benzodiazepines) – Anticonvulsants (e.g., barbiturates) – Antiparkinsonian agents (e.g., benztropine, trihexyphenidyl) . Analgesics – Narcotics (NB. meperidine*) – Non-steroidal anti-inflammatory drugs* . Antihistamines (first generation, e.g., hydroxyzine) . Gastrointestinal agents – Antispasmodics – H2-blockers* . Antinauseants – Scopolamine – Dimenhydrinate . Antibiotics – Fluoroquinolones* . Psychotropic medications – Tricyclic antidepressants – Lithium* . Cardiac medications – Antiarrhythmics – Digitalis* – Antihypertensives (b-blockers, methyldopa) . Miscellaneous – Skeletal muscle relaxants – Steroids Over the counter medications and complementary/alternative medications . Antihistamines (NB. first generation) – diphenhydramine, chlorpheniramine). Antinauseants – dimenhydrinate, scopolamine . Liquid medications containing alcohol . Mandrake . Henbane . Jimson weed . Atropa belladonna extract * Requires adjustment in renal impairment. From: K Alagiakrishnan, C A Wiens. (2004). An approach to drug induced delirium in the elderly. Postgrad Med J, 80, 388–393. Delirium in the Older Person: A Medical Emergency. Island Health www.viha.ca/mhas/resources/delirium/ Drugs that can cause delirium. Reviewed: 8-2014 Some commonly used medications with moderate to high anticholinergic properties and alternative suggestions Type of medication Alternatives with less deliriogenic -

Reorganization Plan Authority” of the Philip Buchen Files at the Gerald R
The original documents are located in Box 59, folder “Reorganization Plan Authority” of the Philip Buchen Files at the Gerald R. Ford Presidential Library. Copyright Notice The copyright law of the United States (Title 17, United States Code) governs the making of photocopies or other reproductions of copyrighted material. Gerald R. Ford donated to the United States of America his copyrights in all of his unpublished writings in National Archives collections. Works prepared by U.S. Government employees as part of their official duties are in the public domain. The copyrights to materials written by other individuals or organizations are presumed to remain with them. If you think any of the information displayed in the PDF is subject to a valid copyright claim, please contact the Gerald R. Ford Presidential Library. Digitized from Box 59 of the Philip Buchen Files at the Gerald R. Ford Presidential Library Monday 3/10/75 7:10 Warren Hendrlcks would like you to take a look at this memo dated 2 /18 from Nichols to Jerry Jonea re extension of Presidential Reorganization Plan authority. I have attached a copy of Mr. Areeda's signoff of 2/21. Apparently nothing has been done and he feels you may not be aware of this • • Monday 3/10/75 7:10 Warren Hendricks would like you to take a look at this memo dated 2 /18 from Nichols to Jerry Jones re extension of Presidential Reorganization Plan authority. I have attached a copy of Mr. Areeda1 s signoff of 2/21. Apparently nothing has been done and he feels you may not be aware of this • • • • , , , ~ A .J...J .n l.J u ::, .L 7 .r\CilO. -
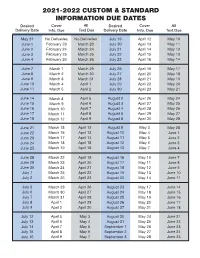
2021-2022 Custom & Standard Information Due Dates
2021-2022 CUSTOM & STANDARD INFORMATION DUE DATES Desired Cover All Desired Cover All Delivery Date Info. Due Text Due Delivery Date Info. Due Text Due May 31 No Deliveries No Deliveries July 19 April 12 May 10 June 1 February 23 March 23 July 20 April 13 May 11 June 2 February 24 March 24 July 21 April 14 May 12 June 3 February 25 March 25 July 22 April 15 May 13 June 4 February 26 March 26 July 23 April 16 May 14 June 7 March 1 March 29 July 26 April 19 May 17 June 8 March 2 March 30 July 27 April 20 May 18 June 9 March 3 March 31 July 28 April 21 May 19 June 10 March 4 April 1 July 29 April 22 May 20 June 11 March 5 April 2 July 30 April 23 May 21 June 14 March 8 April 5 August 2 April 26 May 24 June 15 March 9 April 6 August 3 April 27 May 25 June 16 March 10 April 7 August 4 April 28 May 26 June 17 March 11 April 8 August 5 April 29 May 27 June 18 March 12 April 9 August 6 April 30 May 28 June 21 March 15 April 12 August 9 May 3 May 28 June 22 March 16 April 13 August 10 May 4 June 1 June 23 March 17 April 14 August 11 May 5 June 2 June 24 March 18 April 15 August 12 May 6 June 3 June 25 March 19 April 16 August 13 May 7 June 4 June 28 March 22 April 19 August 16 May 10 June 7 June 29 March 23 April 20 August 17 May 11 June 8 June 30 March 24 April 21 August 18 May 12 June 9 July 1 March 25 April 22 August 19 May 13 June 10 July 2 March 26 April 23 August 20 May 14 June 11 July 5 March 29 April 26 August 23 May 17 June 14 July 6 March 30 April 27 August 24 May 18 June 15 July 7 March 31 April 28 August 25 May 19 June 16 July 8 April 1 April 29 August 26 May 20 June 17 July 9 April 2 April 30 August 27 May 21 June 18 July 12 April 5 May 3 August 30 May 24 June 21 July 13 April 6 May 4 August 31 May 25 June 22 July 14 April 7 May 5 September 1 May 26 June 23 July 15 April 8 May 6 September 2 May 27 June 24 July 16 April 9 May 7 September 3 May 28 June 25. -

2020-2021 Academic Year Grid ALL 11X17
Fall 2020 Spring 2021 Summer 2021* EVENTS / DEADLINES Session 1 Session 1 Session 2 Session 3 Session 4 Session 5 Session 6 Winter Mini Session 2 Session 3 Session 4 Session 5 Session 6 Summer Mini Session 1 Session 2 Session 3 Session 4 Regular Regular First Day of Classes September 28, October 19, November 2, December 21, February 22, *Please also see notes below August 24, 2020 August 24, 2020 August 24, 2020 January 19, 2021 January 19, 2021 January 19, 2021 March 22, 2021 April 5, 2021 May 17, 2021 June 7, 2021 June 7, 2021 June 7, 2021 July 12, 2021 2020 2020 2020 2020 2021 regarding college-specific dates and Monday Monday Monday Tuesday Tuesday Tuesday Monday Monday Monday Monday Monday Monday Monday Monday Monday Monday Monday Monday summer sessions meeting days. Labor Day Holiday (Fall); September 7, 2020 January 18, 2021 May 31, 2021 Martin Luther King Holiday (Spring); Monday Monday Monday Memorial Day (Summer) **Extended** September 30, October 21, November 4, December 22, February 24, Last Day to Add a Class September 1, August 26, 2020 August 26, 2020 January 26, 2021 January 21, 2021 January 21, 2021 March 24, 2021 April 7, 2021 May 18, 2021 June 8, 2021 June 8, 2021 June 8, 2021 July 13, 2021 2020 2020 2020 2020 2021 or be enrolled from the Wait List 2020 Wednesday Wednesday Tuesday Thursday Thursday Wednesday Wednesday Tuesday Tuesday Tuesday Tuesday Tuesday Wednesday Wednesday Wednesday Tuesday Wednesday Tuesday ORD - Official Reporting Day Last day to drop a course or withdraw without receiving a grade Last day to -

Town of Mount Pleasant, South Carolina Finance
TOWN OF MOUNT PLEASANT, SOUTH CAROLINA FINANCE COMMITTEE Tuesday, July 6, 2021 Municipal Complex, Committee Meeting Room – 3rd Floor 100 Ann Edwards Lane, Mount Pleasant, SC 29464 Minutes Members Present: Tom O’Rourke, Chair; Gary Santos, Jake Rambo, Kathy Landing Staff Present: Eric DeMoura, Marcy Cotov 1. Mr. O’Rourke called the meeting to order at 1:35 p.m. 2. Approval of Minutes from the June 1, 2021 meeting Mr. Santos moved to approve; seconded by Ms. Landing. All present voted in favor. 3. Public Comments. None. 4. Discussion and possible action regarding fees for towing service Town of Mount Pleasant Chief of Police, Mark Arnold, presented the following handout. Finance Committee July 6, 2021 Page 2 Finance Committee July 6, 2021 Page 3 Mr. Santos moved to approve; seconded by Mr. Rambo. There was discussion between Committee and staff. All present voted in favor. 5. Discussion and possible action regarding a purchase of property for economic development Mr. O’Rourke stated that this Agenda Item was presented to the Economic Development Committee by Ms. Landing, who agreed during Executive Session to send this item to Council next week. Mr. Rambo moved to go into Executive Session; seconded by Mr. Santos. All present voted in favor. 6. Executive session regarding a purchase of property for economic development Committee went into Executive Session at 1:40 p.m. 7. Post Executive Session Committee may take action on any item, including any subsection of any section, listed on an executive session agenda or discussed in an executive session during a properly noticed meeting.