Comparative Genomic Evidence for the Involvement of Schizophrenia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chuanxiong Rhizoma Compound on HIF-VEGF Pathway and Cerebral Ischemia-Reperfusion Injury’S Biological Network Based on Systematic Pharmacology
ORIGINAL RESEARCH published: 25 June 2021 doi: 10.3389/fphar.2021.601846 Exploring the Regulatory Mechanism of Hedysarum Multijugum Maxim.-Chuanxiong Rhizoma Compound on HIF-VEGF Pathway and Cerebral Ischemia-Reperfusion Injury’s Biological Network Based on Systematic Pharmacology Kailin Yang 1†, Liuting Zeng 1†, Anqi Ge 2†, Yi Chen 1†, Shanshan Wang 1†, Xiaofei Zhu 1,3† and Jinwen Ge 1,4* Edited by: 1 Takashi Sato, Key Laboratory of Hunan Province for Integrated Traditional Chinese and Western Medicine on Prevention and Treatment of 2 Tokyo University of Pharmacy and Life Cardio-Cerebral Diseases, Hunan University of Chinese Medicine, Changsha, China, Galactophore Department, The First 3 Sciences, Japan Hospital of Hunan University of Chinese Medicine, Changsha, China, School of Graduate, Central South University, Changsha, China, 4Shaoyang University, Shaoyang, China Reviewed by: Hui Zhao, Capital Medical University, China Background: Clinical research found that Hedysarum Multijugum Maxim.-Chuanxiong Maria Luisa Del Moral, fi University of Jaén, Spain Rhizoma Compound (HCC) has de nite curative effect on cerebral ischemic diseases, *Correspondence: such as ischemic stroke and cerebral ischemia-reperfusion injury (CIR). However, its Jinwen Ge mechanism for treating cerebral ischemia is still not fully explained. [email protected] †These authors share first authorship Methods: The traditional Chinese medicine related database were utilized to obtain the components of HCC. The Pharmmapper were used to predict HCC’s potential targets. Specialty section: The CIR genes were obtained from Genecards and OMIM and the protein-protein This article was submitted to interaction (PPI) data of HCC’s targets and IS genes were obtained from String Ethnopharmacology, a section of the journal database. -

Downloads/ (Accessed on 17 January 2020)
cells Review Novel Approaches for Identifying the Molecular Background of Schizophrenia Arkadiy K. Golov 1,2,*, Nikolay V. Kondratyev 1 , George P. Kostyuk 3 and Vera E. Golimbet 1 1 Mental Health Research Center, 34 Kashirskoye shosse, 115522 Moscow, Russian; [email protected] (N.V.K.); [email protected] (V.E.G.) 2 Institute of Gene Biology, Russian Academy of Sciences, 34/5 Vavilova Street, 119334 Moscow, Russian 3 Alekseev Psychiatric Clinical Hospital No. 1, 2 Zagorodnoye shosse, 115191 Moscow, Russian; [email protected] * Correspondence: [email protected] Received: 5 November 2019; Accepted: 16 January 2020; Published: 18 January 2020 Abstract: Recent advances in psychiatric genetics have led to the discovery of dozens of genomic loci associated with schizophrenia. However, a gap exists between the detection of genetic associations and understanding the underlying molecular mechanisms. This review describes the basic approaches used in the so-called post-GWAS studies to generate biological interpretation of the existing population genetic data, including both molecular (creation and analysis of knockout animals, exploration of the transcriptional effects of common variants in human brain cells) and computational (fine-mapping of causal variability, gene set enrichment analysis, partitioned heritability analysis) methods. The results of the crucial studies, in which these approaches were used to uncover the molecular and neurobiological basis of the disease, are also reported. Keywords: schizophrenia; GWAS; causal genetic variants; enhancers; brain epigenomics; genome/epigenome editing 1. Introduction Schizophrenia is a severe mental illness that affects between 0.5% and 0.7% of the human population [1]. Both environmental and genetic factors are thought to be involved in its pathogenesis, with genetic factors playing a key role in disease risk, as the heritability of schizophrenia is estimated to be 70–85% [2,3]. -

Efficient Analysis of Mouse Genome Sequences Reveal Many Nonsense Variants
Efficient analysis of mouse genome sequences reveal many nonsense variants Sophie Steelanda,b,1, Steven Timmermansa,b,1, Sara Van Ryckeghema,b, Paco Hulpiaua,b, Yvan Saeysa,c, Marc Van Montagud,e,f,2, Roosmarijn E. Vandenbrouckea,b,3, and Claude Liberta,b,2,3 aInflammation Research Center, Flanders Institute for Biotechnology (VIB), 9052 Ghent, Belgium; bDepartment of Biomedical Molecular Biology, Ghent University, 9052 Ghent, Belgium; cDepartment of Internal Medicine, Ghent University, 9052 Ghent, Belgium; dDepartment of Plant Systems Biology, VIB, 9052 Ghent, Belgium; eDepartment of Plant Biotechnology and Bioinformatics, Ghent University, 9052 Ghent, Belgium; and fInternational Plant Biotechnology Outreach, VIB, Ghent, Belgium Contributed by Marc Van Montagu, March 30, 2016 (sent for review December 31, 2015; reviewed by Bruce Beutler, Stefano Bruscoli, Stefan Rose-John, and Klaus Schulze-Osthoff) Genetic polymorphisms in coding genes play an important role alive, archiving them, and distributing mutant strains to in- when using mouse inbred strains as research models. They have terested users (4). been shown to influence research results, explain phenotypical Since Clarence Little showed in the early 20th century that the differences between inbred strains, and increase the amount of principle of inbreeding also applies to mice, several hundred interesting gene variants present in the many available inbred inbred mouse strains have been generated (5). Some of these lines. SPRET/Ei is an inbred strain derived from Mus spretus that strains display specific phenotypes that are the result of a mutant has ∼1% sequence difference with the C57BL/6J reference ge- gene, and in several cases have formed the basis for identifying nome. -

Network-Based Prediction of Polygenic Disease Genes Involved in Cell Motility Miriam Bern†, Alexander King†, Derek A
Bern et al. BMC Bioinformatics 2019, 20(Suppl 12):313 https://doi.org/10.1186/s12859-019-2834-1 RESEARCH Open Access Network-based prediction of polygenic disease genes involved in cell motility Miriam Bern†, Alexander King†, Derek A. Applewhite and Anna Ritz* From International Workshop on Computational Network Biology: Modeling, Analysis and Control Washington, D.C., USA. 29 August 2018 Abstract Background: Schizophrenia and autism are examples of polygenic diseases caused by a multitude of genetic variants, many of which are still poorly understood. Recently, both diseases have been associated with disrupted neuron motility and migration patterns, suggesting that aberrant cell motility is a phenotype for these neurological diseases. Results: We formulate the POLYGENIC DISEASE PHENOTYPE Problem which seeks to identify candidate disease genes that may be associated with a phenotype such as cell motility. We present a machine learning approach to solve this problem for schizophrenia and autism genes within a brain-specific functional interaction network. Our method outperforms peer semi-supervised learning approaches, achieving better cross-validation accuracy across different sets of gold-standard positives. We identify top candidates for both schizophrenia and autism, and select six genes labeled as schizophrenia positives that are predicted to be associated with cell motility for follow-up experiments. Conclusions: Candidate genes predicted by our method suggest testable hypotheses about these genes’ role in cell motility regulation, offering a framework for generating predictions for experimental validation. Keywords: Semi-supervised learning, Functional interaction network, Schizophrenia, Autism, Cell motility Background participants [4, 5], indicating that schizophrenia arises Many polygenic diseases, which arise from the action from a multitude of cellular perturbations compounding or influence of multiple genes, are difficult to geneti- their effects. -

Research Article a Robust Topology-Based Algorithm for Gene Expression Profiling
International Scholarly Research Network ISRN Bioinformatics Volume 2012, Article ID 381023, 11 pages doi:10.5402/2012/381023 Research Article A Robust Topology-Based Algorithm for Gene Expression Profiling Lars Seemann,1 Jason Shulman,2 and Gemunu H. Gunaratne1 1 Department of Physics, University of Houston, Houston, TX 77204, USA 2 Department of Physics, Richard Stockton College of New Jersey, Pomona, NJ 08240, USA Correspondence should be addressed to Gemunu H. Gunaratne, [email protected] Received 3 August 2012; Accepted 30 August 2012 Academic Editors: B. Haubold and K. Yura Copyright © 2012 Lars Seemann et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Early and accurate diagnoses of cancer can significantly improve the design of personalized therapy and enhance the success of therapeutic interventions. Histopathological approaches, which rely on microscopic examinations of malignant tissue, are not conducive to timely diagnoses. High throughput genomics offers a possible new classification of cancer subtypes. Unfortunately, most clustering algorithms have not been proven sufficiently robust. We propose a novel approach that relies on the use of statistical invariants and persistent homology, one of the most exciting recent developments in topology. It identifies a sufficient but compact set of genes for the analysis as well as a core group of tightly correlated patient samples for each subtype. Partitioning occurs hierarchically and allows for the identification of genetically similar subtypes. We analyzed the gene expression profiles of 202 tumors of the brain cancer glioblastoma multiforme (GBM) given at the Cancer Genome Atlas (TCGA) site. -

(12) United States Patent (10) Patent No.: US 7.873,482 B2 Stefanon Et Al
US007873482B2 (12) United States Patent (10) Patent No.: US 7.873,482 B2 Stefanon et al. (45) Date of Patent: Jan. 18, 2011 (54) DIAGNOSTIC SYSTEM FOR SELECTING 6,358,546 B1 3/2002 Bebiak et al. NUTRITION AND PHARMACOLOGICAL 6,493,641 B1 12/2002 Singh et al. PRODUCTS FOR ANIMALS 6,537,213 B2 3/2003 Dodds (76) Inventors: Bruno Stefanon, via Zilli, 51/A/3, Martignacco (IT) 33035: W. Jean Dodds, 938 Stanford St., Santa Monica, (Continued) CA (US) 90403 FOREIGN PATENT DOCUMENTS (*) Notice: Subject to any disclaimer, the term of this patent is extended or adjusted under 35 WO WO99-67642 A2 12/1999 U.S.C. 154(b) by 158 days. (21)21) Appl. NoNo.: 12/316,8249 (Continued) (65) Prior Publication Data Swanson, et al., “Nutritional Genomics: Implication for Companion Animals'. The American Society for Nutritional Sciences, (2003).J. US 2010/O15301.6 A1 Jun. 17, 2010 Nutr. 133:3033-3040 (18 pages). (51) Int. Cl. (Continued) G06F 9/00 (2006.01) (52) U.S. Cl. ........................................................ 702/19 Primary Examiner—Edward Raymond (58) Field of Classification Search ................... 702/19 (74) Attorney, Agent, or Firm Greenberg Traurig, LLP 702/23, 182–185 See application file for complete search history. (57) ABSTRACT (56) References Cited An analysis of the profile of a non-human animal comprises: U.S. PATENT DOCUMENTS a) providing a genotypic database to the species of the non 3,995,019 A 1 1/1976 Jerome human animal Subject or a selected group of the species; b) 5,691,157 A 1 1/1997 Gong et al. -

In This Table Protein Name, Uniprot Code, Gene Name P-Value
Supplementary Table S1: In this table protein name, uniprot code, gene name p-value and Fold change (FC) for each comparison are shown, for 299 of the 301 significantly regulated proteins found in both comparisons (p-value<0.01, fold change (FC) >+/-0.37) ALS versus control and FTLD-U versus control. Two uncharacterized proteins have been excluded from this list Protein name Uniprot Gene name p value FC FTLD-U p value FC ALS FTLD-U ALS Cytochrome b-c1 complex P14927 UQCRB 1.534E-03 -1.591E+00 6.005E-04 -1.639E+00 subunit 7 NADH dehydrogenase O95182 NDUFA7 4.127E-04 -9.471E-01 3.467E-05 -1.643E+00 [ubiquinone] 1 alpha subcomplex subunit 7 NADH dehydrogenase O43678 NDUFA2 3.230E-04 -9.145E-01 2.113E-04 -1.450E+00 [ubiquinone] 1 alpha subcomplex subunit 2 NADH dehydrogenase O43920 NDUFS5 1.769E-04 -8.829E-01 3.235E-05 -1.007E+00 [ubiquinone] iron-sulfur protein 5 ARF GTPase-activating A0A0C4DGN6 GIT1 1.306E-03 -8.810E-01 1.115E-03 -7.228E-01 protein GIT1 Methylglutaconyl-CoA Q13825 AUH 6.097E-04 -7.666E-01 5.619E-06 -1.178E+00 hydratase, mitochondrial ADP/ATP translocase 1 P12235 SLC25A4 6.068E-03 -6.095E-01 3.595E-04 -1.011E+00 MIC J3QTA6 CHCHD6 1.090E-04 -5.913E-01 2.124E-03 -5.948E-01 MIC J3QTA6 CHCHD6 1.090E-04 -5.913E-01 2.124E-03 -5.948E-01 Protein kinase C and casein Q9BY11 PACSIN1 3.837E-03 -5.863E-01 3.680E-06 -1.824E+00 kinase substrate in neurons protein 1 Tubulin polymerization- O94811 TPPP 6.466E-03 -5.755E-01 6.943E-06 -1.169E+00 promoting protein MIC C9JRZ6 CHCHD3 2.912E-02 -6.187E-01 2.195E-03 -9.781E-01 Mitochondrial 2- -
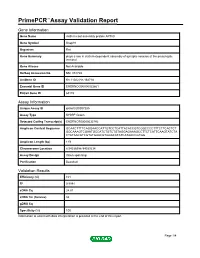
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name clathrin coat assembly protein AP180 Gene Symbol Snap91 Organism Rat Gene Summary plays a role in clathrin-dependent assembly of synaptic vesicles at the presynaptic terminal Gene Aliases Not Available RefSeq Accession No. NM_031728 UniGene ID Rn.11022,Rn.164718 Ensembl Gene ID ENSRNOG00000023861 Entrez Gene ID 65178 Assay Information Unique Assay ID qRnoCID0007285 Assay Type SYBR® Green Detected Coding Transcript(s) ENSRNOT00000032792 Amplicon Context Sequence GCAACTTTTCAGGAACCATTGTCCTCATTACACCGTCGGCCCCTTTCTTCACTCT GGCAAAGTCGAATGCCATCTGTCTGTAGGAGAAAGCCTTCTCATTCAAGTATCTA CTATAACGTCGTATGAACGTGGACATATCATAACCGTGG Amplicon Length (bp) 119 Chromosome Location 8:94036996-94039234 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 101 R2 0.9991 cDNA Cq 24.81 cDNA Tm (Celsius) 82 gDNA Cq Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report Snap91, Rat Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. Detected Coding Transcript(s) This is a list of the Ensembl transcript ID(s) that this assay will detect. -

Effects of Chronic Stress on Prefrontal Cortex Transcriptome in Mice Displaying Different Genetic Backgrounds
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Springer - Publisher Connector J Mol Neurosci (2013) 50:33–57 DOI 10.1007/s12031-012-9850-1 Effects of Chronic Stress on Prefrontal Cortex Transcriptome in Mice Displaying Different Genetic Backgrounds Pawel Lisowski & Marek Wieczorek & Joanna Goscik & Grzegorz R. Juszczak & Adrian M. Stankiewicz & Lech Zwierzchowski & Artur H. Swiergiel Received: 14 May 2012 /Accepted: 25 June 2012 /Published online: 27 July 2012 # The Author(s) 2012. This article is published with open access at Springerlink.com Abstract There is increasing evidence that depression signaling pathway (Clic6, Drd1a,andPpp1r1b). LA derives from the impact of environmental pressure on transcriptome affected by CMS was associated with genetically susceptible individuals. We analyzed the genes involved in behavioral response to stimulus effects of chronic mild stress (CMS) on prefrontal cor- (Fcer1g, Rasd2, S100a8, S100a9, Crhr1, Grm5,and tex transcriptome of two strains of mice bred for high Prkcc), immune effector processes (Fcer1g, Mpo,and (HA)and low (LA) swim stress-induced analgesia that Igh-VJ558), diacylglycerol binding (Rasgrp1, Dgke, differ in basal transcriptomic profiles and depression- Dgkg,andPrkcc), and long-term depression (Crhr1, like behaviors. We found that CMS affected 96 and 92 Grm5,andPrkcc) and/or coding elements of dendrites genes in HA and LA mice, respectively. Among genes (Crmp1, Cntnap4,andPrkcc) and myelin proteins with the same expression pattern in both strains after (Gpm6a, Mal,andMog). The results indicate significant CMS, we observed robust upregulation of Ttr gene contribution of genetic background to differences in coding transthyretin involved in amyloidosis, seizures, stress response gene expression in the mouse prefrontal stroke-like episodes, or dementia. -

Correlated Expression Analysis of Genes Implicated in Schizophrenia
Author’s Accepted Manuscript Correlated expression analysis of genes implicated in schizophrenia: identification of putative disease- related pathways Erin I. Liedtke, Sirey Zhang, John A. Thompson, Stefan Sillau, Judith Gault www.elsevier.com/locate/nhtm PII: S2307-5023(16)30043-1 DOI: http://dx.doi.org/10.1016/j.nhtm.2016.11.002 Reference: NHTM39 To appear in: New Horizons in Translational Medicine Received date: 27 October 2016 Revised date: 23 November 2016 Accepted date: 30 November 2016 Cite this article as: Erin I. Liedtke, Sirey Zhang, John A. Thompson, Stefan Sillau and Judith Gault, Correlated expression analysis of genes implicated in schizophrenia: identification of putative disease-related pathways, New Horizons in Translational Medicine, http://dx.doi.org/10.1016/j.nhtm.2016.11.002 This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting galley proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain. Correlated expression analysis of genes implicated in schizophrenia: identification of putative disease-related pathways Erin I Liedtke1a, Sirey Zhanga1, John A. Thompson, PhDa, Stefan Sillau, PhDb, Judith Gault, PhDa,c* aUniversity of Colorado Denver, Anschutz Medical Campus, Department of Neurosurgery bUniversity of Colorado Denver, Anschutz Medical Campus, Department of Neurology cUniversity of Colorado Denver, Anschutz Medical Campus, Department of Psychiatry *Contact: Judith Gault, PhD 12700 E. -

WO 2016/040794 Al 17 March 2016 (17.03.2016) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/040794 Al 17 March 2016 (17.03.2016) P O P C T (51) International Patent Classification: AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, C12N 1/19 (2006.01) C12Q 1/02 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, C12N 15/81 (2006.01) C07K 14/47 (2006.01) DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, (21) International Application Number: KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, PCT/US20 15/049674 MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (22) International Filing Date: PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, 11 September 2015 ( 11.09.201 5) SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every (26) Publication Language: English kind of regional protection available): ARIPO (BW, GH, (30) Priority Data: GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, 62/050,045 12 September 2014 (12.09.2014) US TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, (71) Applicant: WHITEHEAD INSTITUTE FOR BIOMED¬ DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, ICAL RESEARCH [US/US]; Nine Cambridge Center, LV, MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, Cambridge, Massachusetts 02142-1479 (US). -

Gene Annotation
Gene Annotation Contributions from Carlo Colantuoni and Robert Gentleman What We Are Going To Cover Cells, Genes, Transcripts –> Genomics Experiments Sequence Knowledge Behind Genomics Experiments Annotation of Genes in Genomics Experiments Biological Setup Every cell in the human body contains the entire human genome: 3.3 Gb or ~30K genes. The investigation of gene expression is meaningful because different cells, in different environments, doing different jobs express different genes. Tasks necessary for gene expression analysis: Define what a gene is. Identify genes in a sea of genomic DNA where <3% of DNA is contained in genes. Design and implement probes that will effectively assay expression of ALL (most? many?) genes simultaneously. Cross-reference these probes. 1 Cellular Biology, Gene Expression, and Microarray Analysis DNA RNA Protein Gene: Protein coding unit of genomic DNA with an mRNA intermediate. ~30K genes DNA Sequence is a Necessity Probe START protein coding STOP mRNA AAAAA 5’ UTR 3’ UTR Genomic DNA 3.3 Gb From Genomic DNA to mRNA Transcripts EXONS INTRONS Protein-coding genes are not easy to find - gene density is low, and exons are interrupted by introns. ~30K >30K Alternative splicing Alternative start & stop sites in same RNA molecule RNA editing & SNPs Transcript coverage Homology to other transcripts Hybridization dynamics 3’ bias 2 Unsurpassed as source of expressed sequence Sequence Quality! Redundancy! Completeness? Chaos?!? From Genomic DNA to mRNA Transcripts ~30K >30K >>30K Transcript-Based Gene-Centered Information 3 Design of Gene Expression Probes Content: UniGene, Incyte, Celera Expressed vs. Genomic Source: cDNA libraries, clone collections, oligos Cross-referencing of array probes (across platforms): Sequence <> GenBank <> UniGene <> HomoloGene Possible mis-referencing: Genomic GenBank Acc.#’s Referenced ID has more NT’s than probe Old DB builds DB or table errors – copying and pasting 30K rows in excel … Using RefSeq’s can help.