Complete Mitogenomes of Euploea Mulciber (Nymphalidae: Danainae) and Libythea Celtis (Nymphalidae: Libytheinae) and Their Phylogenetic Implications
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Complete Mitochondrial Genome of the Fall Webworm, Hyphantria
Int. J. Biol. Sci. 2010, 6 172 International Journal of Biological Sciences 2010; 6(2):172-186 © Ivyspring International Publisher. All rights reserved Research Paper The complete mitochondrial genome of the fall webworm, Hyphantria cunea (Lepidoptera: Arctiidae) Fang Liao1,2, Lin Wang3, Song Wu4, Yu-Ping Li4, Lei Zhao1, Guo-Ming Huang2, Chun-Jing Niu2, Yan-Qun Liu4, , Ming-Gang Li1, 1. College of Life Sciences, Nankai University, Tianjin 300071, China 2. Tianjin Entry-Exit Inspection and Quarantine Bureau, Tianjin 300457, China 3. Beijing Entry-Exit Inspection and Quarantine Bureau, Beijing 101113, China 4. College of Bioscience and Biotechnology, Shenyang Agricultural University, Liaoning, Shenyang 110866, China Corresponding author: Y. Q. Liu, College of Bioscience and Biotechnology, Shenyang Agricultural University, Liaoning, Shenyang 110866, China; Tel: 86-24-88487163; E-mail: [email protected]. Or to: M. G. Li, College of Life Sciences, Nankai University, Tianjin 300071, China; Tel: 86-22-23508237; E-mail: [email protected]. Received: 2010.02.03; Accepted: 2010.03.26; Published: 2010.03.29 Abstract The complete mitochondrial genome (mitogenome) of the fall webworm, Hyphantria cunea (Lepidoptera: Arctiidae) was determined. The genome is a circular molecule 15 481 bp long. It presents a typical gene organization and order for completely sequenced lepidopteran mitogenomes, but differs from the insect ancestral type for the placement of tRNAMet. The nucleotide composition of the genome is also highly A + T biased, accounting for 80.38%, with a slightly positive AT skewness (0.010), indicating the occurrence of more As than Ts, as found in the Noctuoidea species. All protein-coding genes (PCGs) are initiated by ATN codons, except for COI, which is tentatively designated by the CGA codon as observed in other le- pidopterans. -

Taxonomic Revision of the Tribe Danaini (Lepidoptera: Nymphalidae: Danainae) from Myanmar
JAPB191_proof ■ 5 February 2017 ■ 1/5 Journal of Asia-Pacific Biodiversity xxx (2017) 1e5 55 HOSTED BY Contents lists available at ScienceDirect 56 57 Journal of Asia-Pacific Biodiversity 58 59 60 journal homepage: http://www.elsevier.com/locate/japb 61 62 63 Original article 64 65 1 Taxonomic revision of the tribe Danaini (Lepidoptera: Nymphalidae: 66 2 67 3 Danainae) from Myanmar 68 4 69 a a a b a,* 5 Q4 Nan Zarchi Win , Eun Young Choi , Jong Bong Choi , Jinyoung Park , Jong Kyun Park 70 6 a 71 7 College of Ecology and Environmental Science, Kyungpook National University, Sangju, Republic of Korea b Department of Nature Survey, National Institute of Ecology, Seocheon, Republic of Korea 72 8 73 9 74 10 article info abstract 75 11 76 12 Article history: The tribe Danaini is reviewed for the first time from Myanmar. Ten species of four genera belonging to 77 13 Received 29 September 2016 two subtribes are taxonomically described. Identification keys for the subtribes, the genera, and all 78 14 Received in revised form species are provided. The adult illustrations for all examined species are also presented. 79 8 November 2016 15 Copyright Ó 2017, National Science Museum of Korea (NSMK) and Korea National Arboretum (KNA). 80 Accepted 11 November 2016 16 Production and hosting by Elsevier. This is an open access article under the CC BY-NC-ND license (http:// Available online xxx 81 17 creativecommons.org/licenses/by-nc-nd/4.0/). 82 18 Keywords: 83 19 butterfly 84 20 Danaini 85 Danainae 21 86 Myanmar 22 87 23 88 24 89 25 Introduction Myanmar is one of the biologically diverse countries in main- 90 26 land Southeast Asia and rich in biodiversity. -

The Complete Mitochondrial Genome of Hipparchia Autonoe (Esper, 1783) (Lepidoptera: Nymphalidae): Investigation of Intraspecific Variations on Mitochondrial Genome
Mitochondrial DNA Part B Resources ISSN: (Print) 2380-2359 (Online) Journal homepage: https://www.tandfonline.com/loi/tmdn20 The complete mitochondrial genome of Hipparchia autonoe (Esper, 1783) (Lepidoptera: Nymphalidae): investigation of intraspecific variations on mitochondrial genome Yeong-Don Lee, Jungmo Lee, Do-Sung Kim, Jonghyun Park, Hong Xi, Jeehee Roh, Dong-Soon Kim, Sang June Nam, Seong-Ki Kim, Jin-Young Song & Jongsun Park To cite this article: Yeong-Don Lee, Jungmo Lee, Do-Sung Kim, Jonghyun Park, Hong Xi, Jeehee Roh, Dong-Soon Kim, Sang June Nam, Seong-Ki Kim, Jin-Young Song & Jongsun Park (2020) The complete mitochondrial genome of Hipparchiaautonoe (Esper, 1783) (Lepidoptera: Nymphalidae): investigation of intraspecific variations on mitochondrial genome, Mitochondrial DNA Part B, 5:2, 1542-1544, DOI: 10.1080/23802359.2020.1742230 To link to this article: https://doi.org/10.1080/23802359.2020.1742230 © 2020 The Author(s). Published by Informa Published online: 24 Mar 2020. UK Limited, trading as Taylor & Francis Group. Submit your article to this journal Article views: 95 View related articles View Crossmark data Full Terms & Conditions of access and use can be found at https://www.tandfonline.com/action/journalInformation?journalCode=tmdn20 MITOCHONDRIAL DNA PART B 2020, VOL. 5, NO. 2, 1542–1544 https://doi.org/10.1080/23802359.2020.1742230 MITOGENOME ANNOUNCEMENT The complete mitochondrial genome of Hipparchia autonoe (Esper, 1783) (Lepidoptera: Nymphalidae): investigation of intraspecific variations on mitochondrial -

Diversity of Takhni Rehmapur Wildlife Sanctuary, Hoshiarpur, Punjab, India
SHARMA : Butterfly Diversity of Takhni Rehmapur Wildlife Sanctuary, Hoshiarpur, Punjab, India 305 ISSN 0375-1511 Rec. zool. Surv. India : 115(Part-3) : 305-311, 2015 BUTTERFLY (LEPIDOPTERA: INSECTA) DIVERSITY OF TAKHNI REHMAPUR WILDLIFE SANCTUARY, HOSHIARPUR, PUNJAB, INDIA NARENDER SHARMA Zoological Survey of India, Northern Regional Centre, 218 Kaulagarh Road, Dehradun-248 195 Email : [email protected] INTRODUCTION the available information is mainly restricted to The butterfly fauna of India has been well that published by Rose and Sidhu (2001), who studied in the past, with the works of Marshall & de provided an inventory of 74 species of butterflies Niceville (1883), de Niceville (1886, 1890), Moore from Punjab; Arora et al. (2006), who gave a brief (1890-1905), Swinhoe (1893, 1896, 1905-1913), account of 74 species from Punajb Shivaliks; and Bingham (1905, 1907), Evans (1932), Talbot Sharma and Joshi (2009), who listed 41 species (1939, 1947), Wynter-Blyth (1957), and Kehimkar from Dholbaha Dam (Hoshiarpur). However, (2008) being some of the significant publications. information on the butterfly diversity of the various To date, 1641 species of butterflies have been protected areas of Punjab is almost totally lacking. reported from India (Varshney, 2010). Recently, It is precisely with this point in mind that while much information on butterflies of different conducting ‘General Faunistic Surveys’ of Punjab regions, states and protected areas of India has under the mandates of the Zoological Survey of been published (e.g. Arora et al. 2009 (Himachal India, we were fortunate to have the opportunity Pradesh); Anonymous (website of Punjab ENVIS to study the butterfly faunal diversity of Takhni Centre, Punjab); Kumar 2008 (Uttarakhand); Rehmapur Wildlife Sanctuary on 12th and 13th Mondal et al., 1997 (Delhi); Chandra et al. -

Development of Encyclopedia Boyong Sleman Insekta River As Alternative Learning Resources
PROC. INTERNAT. CONF. SCI. ENGIN. ISSN 2597-5250 Volume 3, April 2020 | Pages: 629-634 E-ISSN 2598-232X Development of Encyclopedia Boyong Sleman Insekta River as Alternative Learning Resources Rini Dita Fitriani*, Sulistiyawati Biological Education Faculty of Science and Technology, UIN Sunan Kalijaga Jl. Marsda Adisucipto Yogyakarta, Indonesia Email*: [email protected] Abstract. This study aims to determine the types of insects Coleoptera, Hemiptera, Odonata, Orthoptera and Lepidoptera in the Boyong River, Sleman Regency, Yogyakarta, to develop the Encyclopedia of the Boyong River Insect and to determine the quality of the encyclopedia developed. The method used in the research inventory of the types of insects Coleoptera, Hemiptera, Odonata, Orthoptera and Lepidoptera insects in the Boyong River survey method with the results of the study found 46 species of insects consisting of 2 Coleoptera Orders, 2 Hemiptera Orders, 18 orders of Lepidoptera in Boyong River survey method with the results of the research found 46 species of insects consisting of 2 Coleoptera Orders, 2 Hemiptera Orders, 18 orders of Lepidoptera in Boyong River survey method. odonata, 4 Orthopterous Orders and 20 Lepidopterous Orders from 15 families. The encyclopedia that was developed was created using the Adobe Indesig application which was developed in printed form. Testing the quality of the encyclopedia uses a checklist questionnaire and the results of the percentage of ideals from material experts are 91.1% with very good categories, 91.7% of media experts with very good categories, peer reviewers 92.27% with very good categories, biology teachers 88, 53% with a very good category and students 89.8% with a very good category. -
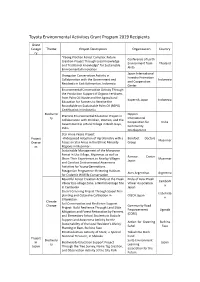
List of Previous Grant Projects
Toyota Environmental Activities Grant Program 2019 Recipients Grant Catego Theme Project Description Organization Country ry "Kaeng Krachan Forest Complex: Future Conference of Earth Creation Project Through Local Knowledge Environment from Thailand and Traditional Knowledge" for Sustainable Akita Environmental Innovation Japan International Orangutan Conservation Activity in Forestry Promotion Collaboration with the Government and Indonesia and Cooperation Residents in East Kalimantan, Indonesia Center Environmental Conservation Activity Through the Production Support of Organic Fertilizers from Palm Oil Waste and the Agricultural Kopernik Japan Indonesia Education for Farmers to Receive the Roundtable on Sustainable Palm Oil (RSPO) Certification in Indonesia Biodiversi Nippon Practical Environmental Education Project in ty International Collaboration with Children, Women, and the Cooperation for India Government in a Rural Village in Bodh Gaya, Community India Development Star Anise Peace Project Project -Widespread Adoption of Agroforestry with a Barefoot Doctors Myanmar Overse Focus on Star Anise in the Ethnic Minority Group as Regions in Myanmar- Sustainable Management of the Mangrove Forest in Uto Village, Myanmar, as well as Ramsar Center Share Their Experiences to Nearby Villages Myanmar Japan and Conduct Environmental Awareness Activities for Young Generations Patagonian Programme: Restoring Habitats Aves Argentinas Argentina for Endemic Wildlife Conservation Beautiful Forest Creation Activity at the Preah Pride of Asia: Preah -

Healthy Planet, Healthy People Guide
HEALTHY PLANET, HEALTHY PEOPLE A Guide to Human Health and Biodiversity UNEP, 2011-2012 declared “Year of the Bat” Bats provide a range of biodiversity benefits to humans … New drug synthesized from vampire bat saliva in development … Could help stroke victims … SAN FRANCISCO, Sep. 6, 2012 … Yosemite National Park … hantavirus warning … 22,000 visitors may have been exposed to deadly mouse-borne disease … confirmed cases growing “Healthy people are better able to learn, be productive and contribute to their communities. At the same time, a healthy environment is a prerequisite for good health.” Dr. Margaret Chan, Director-General of the World Health Organization, 22 June 2012 Healthy Planet Healthy People: A Guide to Human Health and Biodiversity (This page is intentionally left blank) Citation: It is suggested this Guide be cited as: Bridgewater, Peter; Régnier, Mathieu; and Wang Zhen. (2012). HEALTHY PLANET, HEALTHY PEOPLE ‐ A Guide to Human Health and Biodiversity. Secretariat of the Convention on Biological Diversity, Montreal. ACKOWLEDGEMENTS: Alice Barbe, Kathryn Campbell, David Cooper and a number of reviewers who all provided helpful comments and examples. Photo credits (alphabetical order of photographer): P39 bushmeat picture © E.Bennett/WCS, 2009 P47 © Boréalis, 2009 P29, 56, 60, 61 © Peter Bridgewater P39 H5N1 virus © Centre for Disease Control and Prevention, United States of America Government P9, 48, 52 © Christine Estrada P13, 15, 30, 52,58 (marine fresco) © Mathieu Régnier P58 © Urbainculteurs, 2012 The designations employed and the presentation of material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the Convention on Biological Diversity concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. -

Practical Experiences in Invasive Alien Plant Control
ROSALIA Handbooks ROSALIA Handbooks Practical Experiences in Invasive Alien Plant Control Second, revised and expanded edition Invasive plant species pose major agricultural, silvicultural, human health and ecological problems worldwide, and are considered the most signifi cant threat for nature conservation. Species invading natural areas in Hungary have been described by a number of books published in the Practical Experiences in Invasive Alien Plant Control last few years. A great amount of experience has been gathered about the control of these species in some areas, which we can read about in an increasing number of articles; however, no book has been published with regards to the whole country. Invasions affecting larger areas require high energy and cost input, and the effectiveness and successfulness of control can be infl uenced by a number of factors. The development of effective, widely applicable control and eradication technologies is preceded by experiments and examinations which are based on a lot of practical experience and often loaded with negative experiences. National park directorates, forest and agricultural managers and NGOs in many parts of Hungary are combatting the spread of invasive species; however, the exchange of information and conclusion of experiences among the managing bodies is indispensable. The aim of the present volume is to facilitate this by summarizing experiences and the methods applied in practice; which, we hope, will enable us to successfully stop the further spread of invasive plant species and effectively protect our natural values. Magyarország-Szlovákia Partnerséget építünk Határon Átnyúló Együttműködési Program 2007-2013 Duna-Ipoly National Park Directorate rrosaliaosalia kkezikonyvezikonyv 3 aangng jjav.inddav.indd 1 22017.12.15.017.12.15. -

INSECTA: LEPIDOPTERA) DE GUATEMALA CON UNA RESEÑA HISTÓRICA Towards a Synthesis of the Papilionoidea (Insecta: Lepidoptera) from Guatemala with a Historical Sketch
ZOOLOGÍA-TAXONOMÍA www.unal.edu.co/icn/publicaciones/caldasia.htm Caldasia 31(2):407-440. 2009 HACIA UNA SÍNTESIS DE LOS PAPILIONOIDEA (INSECTA: LEPIDOPTERA) DE GUATEMALA CON UNA RESEÑA HISTÓRICA Towards a synthesis of the Papilionoidea (Insecta: Lepidoptera) from Guatemala with a historical sketch JOSÉ LUIS SALINAS-GUTIÉRREZ El Colegio de la Frontera Sur (ECOSUR). Unidad Chetumal. Av. Centenario km. 5.5, A. P. 424, C. P. 77900. Chetumal, Quintana Roo, México, México. [email protected] CLAUDIO MÉNDEZ Escuela de Biología, Universidad de San Carlos, Ciudad Universitaria, Campus Central USAC, Zona 12. Guatemala, Guatemala. [email protected] MERCEDES BARRIOS Centro de Estudios Conservacionistas (CECON), Universidad de San Carlos, Avenida La Reforma 0-53, Zona 10, Guatemala, Guatemala. [email protected] CARMEN POZO El Colegio de la Frontera Sur (ECOSUR). Unidad Chetumal. Av. Centenario km. 5.5, A. P. 424, C. P. 77900. Chetumal, Quintana Roo, México, México. [email protected] JORGE LLORENTE-BOUSQUETS Museo de Zoología, Facultad de Ciencias, UNAM. Apartado Postal 70-399, México D.F. 04510; México. [email protected]. Autor responsable. RESUMEN La riqueza biológica de Mesoamérica es enorme. Dentro de esta gran área geográfi ca se encuentran algunos de los ecosistemas más diversos del planeta (selvas tropicales), así como varios de los principales centros de endemismo en el mundo (bosques nublados). Países como Guatemala, en esta gran área biogeográfi ca, tiene grandes zonas de bosque húmedo tropical y bosque mesófi lo, por esta razón es muy importante para analizar la diversidad en la región. Lamentablemente, la fauna de mariposas de Guatemala es poco conocida y por lo tanto, es necesario llevar a cabo un estudio y análisis de la composición y la diversidad de las mariposas (Lepidoptera: Papilionoidea) en Guatemala. -

Photonic Crystal Structure of Wing Scales in Sasakia Charonda Butterflies
Materials Transactions, Vol. 51, No. 2 (2010) pp. 202 to 208 Special Issue on Development and Fabrication of Advanced Materials Assisted by Nanotechnology and Microanalysis #2010 The Japan Institute of Metals Photonic Crystal Structure of Wing Scales in Sasakia Charonda Butterflies Jirˇina Mateˇjkova´-Plsˇkova´1, Dalibor Jancˇik1, Miroslav Masˇla´nˇ1, Satoshi Shiojiri2 and Makoto Shiojiri3;* 1Centre for Nanomaterial Research, Faculty of Science, Palacky University in Olomouc, Slechtitelu 11, 783 71 Olomouc, Czech Republic 2Matsubara Junior High-school, Kyoto 604-8812, Japan 3Professor Emeritus of Kyoto Institute of Technology, 1-297 Wakiyama, Kyoto 618-0091, Japan The hindwings of the male Sasakia charonda charonda butterflies comprise iridescent purple-blue areas, iridescent white-pearl areas, yellow spots and red spots as well as brown background. We have examined the microstructure of their scales by scanning electron microscopy, for applying their photonic crystal structures to fine light manipulators such as reflection elements in laser diodes. The scales in the yellow spots, red spots and brown background have almost the same structure, which is an optical diffraction grating made of ridges with two cuticle layers. Their difference comes from the contained pigments. The scales in the iridescent purple-blue and white-pearl are also the same in structure. They have seven tilted cuticle layers lapped on the ridges, which also constitute a grating. The widths of the ridge and groove in the grating are different between scales of the two kinds. It is shown that the vivid iridescence is mainly attributed to multiple interferences caused between rays reflected from the seven cuticle layers with air gaps. -

BUTTERFLIES in Thewest Indies of the Caribbean
PO Box 9021, Wilmington, DE 19809, USA E-mail: [email protected]@focusonnature.com Phone: Toll-free in USA 1-888-721-3555 oror 302/529-1876302/529-1876 BUTTERFLIES and MOTHS in the West Indies of the Caribbean in Antigua and Barbuda the Bahamas Barbados the Cayman Islands Cuba Dominica the Dominican Republic Guadeloupe Jamaica Montserrat Puerto Rico Saint Lucia Saint Vincent the Virgin Islands and the ABC islands of Aruba, Bonaire, and Curacao Butterflies in the Caribbean exclusively in Trinidad & Tobago are not in this list. Focus On Nature Tours in the Caribbean have been in: January, February, March, April, May, July, and December. Upper right photo: a HISPANIOLAN KING, Anetia jaegeri, photographed during the FONT tour in the Dominican Republic in February 2012. The genus is nearly entirely in West Indian islands, the species is nearly restricted to Hispaniola. This list of Butterflies of the West Indies compiled by Armas Hill Among the butterfly groupings in this list, links to: Swallowtails: family PAPILIONIDAE with the genera: Battus, Papilio, Parides Whites, Yellows, Sulphurs: family PIERIDAE Mimic-whites: subfamily DISMORPHIINAE with the genus: Dismorphia Subfamily PIERINAE withwith thethe genera:genera: Ascia,Ascia, Ganyra,Ganyra, Glutophrissa,Glutophrissa, MeleteMelete Subfamily COLIADINAE with the genera: Abaeis, Anteos, Aphrissa, Eurema, Kricogonia, Nathalis, Phoebis, Pyrisitia, Zerene Gossamer Wings: family LYCAENIDAE Hairstreaks: subfamily THECLINAE with the genera: Allosmaitia, Calycopis, Chlorostrymon, Cyanophrys, -

Mt Mabu, Mozambique: Biodiversity and Conservation
Darwin Initiative Award 15/036: Monitoring and Managing Biodiversity Loss in South-East Africa's Montane Ecosystems MT MABU, MOZAMBIQUE: BIODIVERSITY AND CONSERVATION November 2012 Jonathan Timberlake, Julian Bayliss, Françoise Dowsett-Lemaire, Colin Congdon, Bill Branch, Steve Collins, Michael Curran, Robert J. Dowsett, Lincoln Fishpool, Jorge Francisco, Tim Harris, Mirjam Kopp & Camila de Sousa ABRI african butterfly research in Forestry Research Institute of Malawi Biodiversity of Mt Mabu, Mozambique, page 2 Front cover: Main camp in lower forest area on Mt Mabu (JB). Frontispiece: View over Mabu forest to north (TT, top); Hermenegildo Matimele plant collecting (TT, middle L); view of Mt Mabu from abandoned tea estate (JT, middle R); butterflies (Lachnoptera ayresii) mating (JB, bottom L); Atheris mabuensis (JB, bottom R). Photo credits: JB – Julian Bayliss CS ‒ Camila de Sousa JT – Jonathan Timberlake TT – Tom Timberlake TH – Tim Harris Suggested citation: Timberlake, J.R., Bayliss, J., Dowsett-Lemaire, F., Congdon, C., Branch, W.R., Collins, S., Curran, M., Dowsett, R.J., Fishpool, L., Francisco, J., Harris, T., Kopp, M. & de Sousa, C. (2012). Mt Mabu, Mozambique: Biodiversity and Conservation. Report produced under the Darwin Initiative Award 15/036. Royal Botanic Gardens, Kew, London. 94 pp. Biodiversity of Mt Mabu, Mozambique, page 3 LIST OF CONTENTS List of Contents .......................................................................................................................... 3 List of Tables .............................................................................................................................