Phylogenomics Reveals Major Diversification Rate Shifts in The
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Moths Light a Way? by John Pickering, Tori Staples and Rebecca Walcott
SOUTHERN LEPIDOPTERISTS NEWS VOLUME 38 NO4. (2016), PG. 331 SAVE ALL SPECIES – MOTHS LIGHT A WAY? BY JOHN PICKERING, TORI STAPLES AND REBECCA WALCOTT Abstract -- What would it take to save all species from snakes, and stinging insects, they pose no health risk. extinction? A new initiative, Save all species, plans to Moths are an exceedingly species-rich group, for which answer this question and provide the tools we need to do the diversity at a terrestrial site will typically exceed any so by 2050. Here we consider the merits and problems other taxon except for beetles. Because moth larvae are associated with inventorying moths to help decide which restricted in their diet to specific host taxa, differences in terrestrial areas to protect. We compare the the assemblages of resident moth species could reflect scientifically-described moth fauna of the British Isles differences across sites in plants and other hosts. If which, with 2,441 species, is taxonomically complete, that’s true, we may be able to use moth inventories as with 11,806 described species from North America north efficient proxies to compare surrounding plant of Mexico, the fauna of which is not fully described. As communities. a percentage of the described moth fauna, there are fewer “macro” moths (Geometroidea, Drepanoidea, Inventorying moths presents challenges, notably, Noctuoidea, Bombycoidea, Lasiocampidae) in the sampling smaller species, describing thousands of British Isles (34.9%) than those known for the United species new to science, and identifying specimens States and Canada (46.1%). We present data on 1,254 accurately. Our experience is that we can identify 99% species for an intensively-studied site in Clarke County, of moths from digital images to species, species-groups, Georgia and consider whether species in the British Isles which contain species of similar appearance, or are generally smaller than ones in Georgia. -

Modular Structure, Sequence Diversification and Appropriate
www.nature.com/scientificreports OPEN Modular structure, sequence diversifcation and appropriate nomenclature of seroins produced Received: 17 July 2018 Accepted: 14 February 2019 in the silk glands of Lepidoptera Published: xx xx xxxx Lucie Kucerova1, Michal Zurovec 1,2, Barbara Kludkiewicz1, Miluse Hradilova3, Hynek Strnad3 & Frantisek Sehnal1,2 Seroins are small lepidopteran silk proteins known to possess antimicrobial activities. Several seroin paralogs and isoforms were identifed in studied lepidopteran species and their classifcation required detailed phylogenetic analysis based on complete and verifed cDNA sequences. We sequenced silk gland-specifc cDNA libraries from ten species and identifed 52 novel seroin cDNAs. The results of this targeted research, combined with data retrieved from available databases, form a dataset representing the major clades of Lepidoptera. The analysis of deduced seroin proteins distinguished three seroin classes (sn1-sn3), which are composed of modules: A (includes the signal peptide), B (rich in charged amino acids) and C (highly variable linker containing proline). The similarities within and between the classes were 31–50% and 22.5–25%, respectively. All species express one, and in exceptional cases two, genes per class, and alternative splicing further enhances seroin diversity. Seroins occur in long versions with the full set of modules (AB1C1B2C2B3) and/or in short versions that lack parts or the entire B and C modules. The classes and the modular structure of seroins probably evolved prior to the split between Trichoptera and Lepidoptera. The diversity of seroins is refected in proposed nomenclature. Te silk spun by caterpillars is a composite material based on two protein agglomerates that have been known for centuries as fbroin and sericin. -

Lepidoptera of North America 5
Lepidoptera of North America 5. Contributions to the Knowledge of Southern West Virginia Lepidoptera Contributions of the C.P. Gillette Museum of Arthropod Diversity Colorado State University Lepidoptera of North America 5. Contributions to the Knowledge of Southern West Virginia Lepidoptera by Valerio Albu, 1411 E. Sweetbriar Drive Fresno, CA 93720 and Eric Metzler, 1241 Kildale Square North Columbus, OH 43229 April 30, 2004 Contributions of the C.P. Gillette Museum of Arthropod Diversity Colorado State University Cover illustration: Blueberry Sphinx (Paonias astylus (Drury)], an eastern endemic. Photo by Valeriu Albu. ISBN 1084-8819 This publication and others in the series may be ordered from the C.P. Gillette Museum of Arthropod Diversity, Department of Bioagricultural Sciences and Pest Management Colorado State University, Fort Collins, CO 80523 Abstract A list of 1531 species ofLepidoptera is presented, collected over 15 years (1988 to 2002), in eleven southern West Virginia counties. A variety of collecting methods was used, including netting, light attracting, light trapping and pheromone trapping. The specimens were identified by the currently available pictorial sources and determination keys. Many were also sent to specialists for confirmation or identification. The majority of the data was from Kanawha County, reflecting the area of more intensive sampling effort by the senior author. This imbalance of data between Kanawha County and other counties should even out with further sampling of the area. Key Words: Appalachian Mountains, -

United States National Museum Bulletin 276
,*f»W*»"*^W»i;|. SMITHSONIAN INSTITUTION MUSEUM O F NATURAL HISTORY UNITED STATES NATIONAL MUSEUM BULLETIN 276 A Revision of the Genus Malacosoma Hlibner in North America (Lepidoptera: Lasiocampidae): Systematics, Biology, Immatures, and Parasites FREDERICK W. STEHR and EDWIN F. COOK SMITHSONIAN INSTITUTION PRESS CITY OF WASHINGTON 1968 PUBLICATIONS OF THE UNITED STATES NATIONAL MUSEUM The scientific publications of the United States National Museum include two series. Proceedings of the United States National Museum and United States National Museum Bulletin. In these series are published original articles and monographs dealing with the collections and work of the Museum and setting forth newly acquired facts in the field of anthropology, biology, geology, history, and technology. Copies of each publication are distributed to libraries and scientific organizations and to specialists and others interested in the various subjects. The Proceedings, begun in 1878, are intended for the publication, in separate form, of shorter papers. These are gathered in volumes, octavo in size, with the publication date of each paper recorded in the table of contents of the volume. In the Bulletin series, the first of which was issued in 1875, appear longer, separate publications consisting of monographs (occasionally in several parts) and volumes in which are collected works on related subjects. Bulletins are either octavo or quarto in size, depending on the needs of the presentation. Since 1902, papers relating to the botanical collections of the Museum have been published in the Bulletin series under the heading Contributions from the United States National Herbarium. This work forms number 276 of the Bulletin series. -

Butterflies and Moths of Gwinnett County, Georgia, United States
Heliothis ononis Flax Bollworm Moth Coptotriche aenea Blackberry Leafminer Argyresthia canadensis Apyrrothrix araxes Dull Firetip Phocides pigmalion Mangrove Skipper Phocides belus Belus Skipper Phocides palemon Guava Skipper Phocides urania Urania skipper Proteides mercurius Mercurial Skipper Epargyreus zestos Zestos Skipper Epargyreus clarus Silver-spotted Skipper Epargyreus spanna Hispaniolan Silverdrop Epargyreus exadeus Broken Silverdrop Polygonus leo Hammock Skipper Polygonus savigny Manuel's Skipper Chioides albofasciatus White-striped Longtail Chioides zilpa Zilpa Longtail Chioides ixion Hispaniolan Longtail Aguna asander Gold-spotted Aguna Aguna claxon Emerald Aguna Aguna metophis Tailed Aguna Typhedanus undulatus Mottled Longtail Typhedanus ampyx Gold-tufted Skipper Polythrix octomaculata Eight-spotted Longtail Polythrix mexicanus Mexican Longtail Polythrix asine Asine Longtail Polythrix caunus (Herrich-Schäffer, 1869) Zestusa dorus Short-tailed Skipper Codatractus carlos Carlos' Mottled-Skipper Codatractus alcaeus White-crescent Longtail Codatractus yucatanus Yucatan Mottled-Skipper Codatractus arizonensis Arizona Skipper Codatractus valeriana Valeriana Skipper Urbanus proteus Long-tailed Skipper Urbanus viterboana Bluish Longtail Urbanus belli Double-striped Longtail Urbanus pronus Pronus Longtail Urbanus esmeraldus Esmeralda Longtail Urbanus evona Turquoise Longtail Urbanus dorantes Dorantes Longtail Urbanus teleus Teleus Longtail Urbanus tanna Tanna Longtail Urbanus simplicius Plain Longtail Urbanus procne Brown Longtail -

Hatay Yellow Strain (Bombyx Mori L.) Under the Threat of Extinction: Determination of Morphological and Biological Features with Some Reviews
Mustafa Kemal Üniversitesi Tarım Bilimleri Dergisi 26 (2):266-271, 2021 (Mustafa Kemal University Journal of Agricultural Sciences 26 (2):266-271, 2021) e-ISSN: 2667-7733 http://dergipark.org.tr/mkutbd ARAŞTIRMA MAKALESİ RESEARCH ARTICLE Hatay yellow strain (Bombyx mori L.) under the threat of extinction: Determination of morphological and biological features with some reviews İpekböcekçiliğinde yok olma tehditi altında olan Hatay sarısı ırkı (Bombyx mori L.): Bazı incelemelerle morfolojik ve biyolojik özelliklerinin belirlenmesi Başak ULAŞLI1 , Berna İLERİ2 , Feza CAN1 1Hatay Mustafa Kemal University, Faculty of Agriculture, Department of Plant Protect, Antakya-Hatay, Turkey. 2Canakkale Onsekiz Mart University, Faculty of Fine Arts, Department of Textile and Fashion Design, Çanakkale, Turkey. MAKALE BİLGİSİ / ARTICLE INFO Ö Z E T / A B S T R A C T Makale tarihçesi / Article history: Aims: Throughout the history silk fiber is one of the indispensable and DOI: 10.37908/mkutbd.860085 most valuable textile products. Hatay yellow strain, is an indigenous to Geliş tarihi /Received:13.01.2021 Turkey, was domesticated about 5000 years ago and it’s a privilege to have Kabul tarihi/Accepted:04.03.2021 this moth species in our country. In sericulture, the only hibrid strain that weave white coccon has been bred for many years in our country. Keywords: Bombyx mori, Hatay yellow However, Hatay yellow hasn’t been bred for nearly 45 years. In this study, strain, peace silk, biology, Turkey. it is aimed to investigate some morphological and biological characteristics of the Hatay yellow which is facing the danger of extinction. Corresponding author: Başak ULAŞLI Methods and Results: The development of 1000 first-stage larvae with : [email protected] hatching from the eggs in March, which are cultured in the natural habitat under the climate-appropriate lighting time, temperature and humidity conditions in Harbiye district of Hatay province in 2020; the period of five larvae stages, molting four times, coccon period and moth emergence from the cocoons were observed. -

National Parks Association of the Australian Capital Territory Inc
Volume 53 Number 2 June 2016 National Parks Association of the Australian Capital Territory Inc. Burning Aranda Bushland Canberra Nature Map Jagungal Wilderness NPA Bulletin Volume 53 number 2 June 2016 Articles by contributors may not necessarily reflect association opinion or objectives. CONTENTS NPA outings program, June – September 2016 ...............13–16 From the Committee ................................................................2 Bushwalks Rod Griffiths and Christine Goonrey Exciting Rendezvous Valley pack walk ..........................17 The vital work of the National Parks Australia Council ..........3 Esther Gallant Rod Griffiths Mount Tantangara ...........................................................18 NPA's Nature Play program .....................................................3 Brian Slee Graham Scully Pretty Plain ......................................................................19 Aranda Bushland's recent hazard-reduction burn ....................4 Brian Slee Judy Kelly, with Michael Doherty and John Brickhill Glenburn Precinct news..........................................................20 Obituaries .................................................................................6 Col McAlister Book reviews. Leaf Litter, exploring the Mysteries................21 The National Rock Garden ......................................................7 of a Hidden World by Rachel Tonkin Compiled by Kevin McCue Judy Kelly Stolen .......................................................................................7 -

ILLUSTRATIONS of MOTHS in TAIWAN, 1-5 by B. S. Chang
142 BOOK REVIEW TROPICAL LEPIDOPTERJ Tropical Lepidoptera, 4(2): 142 BOOK REVIEW ILLUSTRATIONS OF MOTHS IN TAIWAN, 1-5 by B. S. Chang Vol. 1: Sphingidae, Ratardidae. Epipyropidae, Drepanidae, Cyclidiidae, Thyatiridae, Epicopeiidae, Callidulidae. Lasiocampidae. EupterotidJ Bombycidae, Brahmaeidae, Agaristidae [Noctuidae, part]. 242 pp, 271 col. fig., col. cover. 1989. (paper only) Vol. 2: Arctiidae, Hypsidae [Noctuidae, part], Limacodidae, Notodontidae. 310 pp, 356 col. fig., col. cover. 1989. Vol. 3: Geometridae [1]. Oenochrominae, Geometrinae, Sterrhinae, Larentiinae. 350 pp, 405 col. fig., col. cover. 1989. Vol. 4: Geometridae [2]. Ennominae. 480 pp, 684 col. fig., col. cover. 1990. Vol. 5: Noctuidae [1]. 366 pp. 572 col. fig., col. cover. 1991. (paper only) 1989-91. Taiwan Museum, Taipei. All are 21 x 18.5 cm. (elongate) In Chinese; Latin names. Price for each is S35.00 paper, S42.00 cloth. Available from Flora & Fauna Books, P. O. Box 15718, Gainesville, FL 32604 (plus S2 shipping each, or 55 per set). This series of small, full-color books is the result of a lifetime of with 706 species illustrated out of the 791 recorded for Taiwan. TlJ collecting by retired Taiwan high school teacher B. S. Chang. Until color figures are all excellent but many of the wing venation drawing 1991, 5 volumes were completed, but the second part of the Noctuidae are very faintly reproduced in the books. has unfortunately been halted by the untimely death of the author. It is Overall, the books are one of the unique treatments of Lepidoptera ii not known if a manuscript is available for the eventual completion of the world. -

The Mcguire Center for Lepidoptera and Biodiversity
Supplemental Information All specimens used within this study are housed in: the McGuire Center for Lepidoptera and Biodiversity (MGCL) at the Florida Museum of Natural History, Gainesville, USA (FLMNH); the University of Maryland, College Park, USA (UMD); the Muséum national d’Histoire naturelle in Paris, France (MNHN); and the Australian National Insect Collection in Canberra, Australia (ANIC). Methods DNA extraction protocol of dried museum specimens (detailed instructions) Prior to tissue sampling, dried (pinned or papered) specimens were assigned MGCL barcodes, photographed, and their labels digitized. Abdomens were then removed using sterile forceps, cleaned with 100% ethanol between each sample, and the remaining specimens were returned to their respective trays within the MGCL collections. Abdomens were placed in 1.5 mL microcentrifuge tubes with the apex of the abdomen in the conical end of the tube. For larger abdomens, 5 mL microcentrifuge tubes or larger were utilized. A solution of proteinase K (Qiagen Cat #19133) and genomic lysis buffer (OmniPrep Genomic DNA Extraction Kit) in a 1:50 ratio was added to each abdomen containing tube, sufficient to cover the abdomen (typically either 300 µL or 500 µL) - similar to the concept used in Hundsdoerfer & Kitching (1). Ratios of 1:10 and 1:25 were utilized for low quality or rare specimens. Low quality specimens were defined as having little visible tissue inside of the abdomen, mold/fungi growth, or smell of bacterial decay. Samples were incubated overnight (12-18 hours) in a dry air oven at 56°C. Importantly, we also adjusted the ratio depending on the tissue type, i.e., increasing the ratio for particularly large or egg-containing abdomens. -
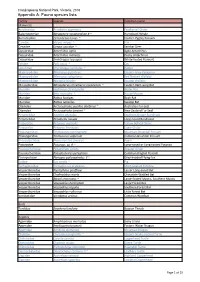
Report-VIC-Croajingolong National Park-Appendix A
Croajingolong National Park, Victoria, 2016 Appendix A: Fauna species lists Family Species Common name Mammals Acrobatidae Acrobates pygmaeus Feathertail Glider Balaenopteriae Megaptera novaeangliae # ~ Humpback Whale Burramyidae Cercartetus nanus ~ Eastern Pygmy Possum Canidae Vulpes vulpes ^ Fox Cervidae Cervus unicolor ^ Sambar Deer Dasyuridae Antechinus agilis Agile Antechinus Dasyuridae Antechinus mimetes Dusky Antechinus Dasyuridae Sminthopsis leucopus White-footed Dunnart Felidae Felis catus ^ Cat Leporidae Oryctolagus cuniculus ^ Rabbit Macropodidae Macropus giganteus Eastern Grey Kangaroo Macropodidae Macropus rufogriseus Red Necked Wallaby Macropodidae Wallabia bicolor Swamp Wallaby Miniopteridae Miniopterus schreibersii oceanensis ~ Eastern Bent-wing Bat Muridae Hydromys chrysogaster Water Rat Muridae Mus musculus ^ House Mouse Muridae Rattus fuscipes Bush Rat Muridae Rattus lutreolus Swamp Rat Otariidae Arctocephalus pusillus doriferus ~ Australian Fur-seal Otariidae Arctocephalus forsteri ~ New Zealand Fur Seal Peramelidae Isoodon obesulus Southern Brown Bandicoot Peramelidae Perameles nasuta Long-nosed Bandicoot Petauridae Petaurus australis Yellow Bellied Glider Petauridae Petaurus breviceps Sugar Glider Phalangeridae Trichosurus cunninghami Mountain Brushtail Possum Phalangeridae Trichosurus vulpecula Common Brushtail Possum Phascolarctidae Phascolarctos cinereus Koala Potoroidae Potorous sp. # ~ Long-nosed or Long-footed Potoroo Pseudocheiridae Petauroides volans Greater Glider Pseudocheiridae Pseudocheirus peregrinus -

Phylogeny and Evolution of Lepidoptera
EN62CH15-Mitter ARI 5 November 2016 12:1 I Review in Advance first posted online V E W E on November 16, 2016. (Changes may R S still occur before final publication online and in print.) I E N C N A D V A Phylogeny and Evolution of Lepidoptera Charles Mitter,1,∗ Donald R. Davis,2 and Michael P. Cummings3 1Department of Entomology, University of Maryland, College Park, Maryland 20742; email: [email protected] 2Department of Entomology, National Museum of Natural History, Smithsonian Institution, Washington, DC 20560 3Laboratory of Molecular Evolution, Center for Bioinformatics and Computational Biology, University of Maryland, College Park, Maryland 20742 Annu. Rev. Entomol. 2017. 62:265–83 Keywords Annu. Rev. Entomol. 2017.62. Downloaded from www.annualreviews.org The Annual Review of Entomology is online at Hexapoda, insect, systematics, classification, butterfly, moth, molecular ento.annualreviews.org systematics This article’s doi: Access provided by University of Maryland - College Park on 11/20/16. For personal use only. 10.1146/annurev-ento-031616-035125 Abstract Copyright c 2017 by Annual Reviews. Until recently, deep-level phylogeny in Lepidoptera, the largest single ra- All rights reserved diation of plant-feeding insects, was very poorly understood. Over the past ∗ Corresponding author two decades, building on a preceding era of morphological cladistic stud- ies, molecular data have yielded robust initial estimates of relationships both within and among the ∼43 superfamilies, with unsolved problems now yield- ing to much larger data sets from high-throughput sequencing. Here we summarize progress on lepidopteran phylogeny since 1975, emphasizing the superfamily level, and discuss some resulting advances in our understanding of lepidopteran evolution. -

Amphiesmeno- Ptera: the Caddisflies and Lepidoptera
CY501-C13[548-606].qxd 2/16/05 12:17 AM Page 548 quark11 27B:CY501:Chapters:Chapter-13: 13Amphiesmeno-Amphiesmenoptera: The ptera:Caddisflies The and Lepidoptera With very few exceptions the life histories of the orders Tri- from Old English traveling cadice men, who pinned bits of choptera (caddisflies)Caddisflies and Lepidoptera (moths and butter- cloth to their and coats to advertise their fabrics. A few species flies) are extremely different; the former have aquatic larvae, actually have terrestrial larvae, but even these are relegated to and the latter nearly always have terrestrial, plant-feeding wet leaf litter, so many defining features of the order concern caterpillars. Nonetheless, the close relationship of these two larval adaptations for an almost wholly aquatic lifestyle (Wig- orders hasLepidoptera essentially never been disputed and is supported gins, 1977, 1996). For example, larvae are apneustic (without by strong morphological (Kristensen, 1975, 1991), molecular spiracles) and respire through a thin, permeable cuticle, (Wheeler et al., 2001; Whiting, 2002), and paleontological evi- some of which have filamentous abdominal gills that are sim- dence. Synapomorphies linking these two orders include het- ple or intricately branched (Figure 13.3). Antennae and the erogametic females; a pair of glands on sternite V (found in tentorium of larvae are reduced, though functional signifi- Trichoptera and in basal moths); dense, long setae on the cance of these features is unknown. Larvae do not have pro- wing membrane (which are modified into scales in Lepi- legs on most abdominal segments, save for a pair of anal pro- doptera); forewing with the anal veins looping up to form a legs that have sclerotized hooks for anchoring the larva in its double “Y” configuration; larva with a fused hypopharynx case.