The Mcguire Center for Lepidoptera and Biodiversity
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Lepidoptera Sphingidae:) of the Caatinga of Northeast Brazil: a Case Study in the State of Rio Grande Do Norte
212212 JOURNAL OF THE LEPIDOPTERISTS’ SOCIETY Journal of the Lepidopterists’ Society 59(4), 2005, 212–218 THE HIGHLY SEASONAL HAWKMOTH FAUNA (LEPIDOPTERA SPHINGIDAE:) OF THE CAATINGA OF NORTHEAST BRAZIL: A CASE STUDY IN THE STATE OF RIO GRANDE DO NORTE JOSÉ ARAÚJO DUARTE JÚNIOR Programa de Pós-Graduação em Ciências Biológicas, Departamento de Sistemática e Ecologia, Universidade Federal da Paraíba, 58059-900, João Pessoa, Paraíba, Brasil. E-mail: [email protected] AND CLEMENS SCHLINDWEIN Departamento de Botânica, Universidade Federal de Pernambuco, Av. Prof. Moraes Rego, s/n, Cidade Universitária, 50670-901, Recife, Pernambuco, Brasil. E-mail:[email protected] ABSTRACT: The caatinga, a thorn-shrub succulent savannah, is located in Northeastern Brazil and characterized by a short and irregular rainy season and a severe dry season. Insects are only abundant during the rainy months, displaying a strong seasonal pat- tern. Here we present data from a yearlong Sphingidae survey undertaken in the reserve Estação Ecológica do Seridó, located in the state of Rio Grande do Norte. Hawkmoths were collected once a month during two subsequent new moon nights, between 18.00h and 05.00h, attracted with a 160-watt mercury vapor light. A total of 593 specimens belonging to 20 species and 14 genera were col- lected. Neogene dynaeus, Callionima grisescens, and Hyles euphorbiarum were the most abundant species, together comprising up to 82.2% of the total number of specimens collected. These frequent species are residents of the caatinga of Rio Grande do Norte. The rare Sphingidae in this study, Pseudosphinx tetrio, Isognathus australis, and Cocytius antaeus, are migratory species for the caatinga. -

The Sphingidae (Lepidoptera) of the Philippines
©Entomologischer Verein Apollo e.V. Frankfurt am Main; download unter www.zobodat.at Nachr. entomol. Ver. Apollo, Suppl. 17: 17-132 (1998) 17 The Sphingidae (Lepidoptera) of the Philippines Willem H o g e n e s and Colin G. T r e a d a w a y Willem Hogenes, Zoologisch Museum Amsterdam, Afd. Entomologie, Plantage Middenlaan 64, NL-1018 DH Amsterdam, The Netherlands Colin G. T readaway, Entomologie II, Forschungsinstitut Senckenberg, Senckenberganlage 25, D-60325 Frankfurt am Main, Germany Abstract: This publication covers all Sphingidae known from the Philippines at this time in the form of an annotated checklist. (A concise checklist of the species can be found in Table 4, page 120.) Distribution maps are included as well as 18 colour plates covering all but one species. Where no specimens of a particular spe cies from the Philippines were available to us, illustrations are given of specimens from outside the Philippines. In total we have listed 117 species (with 5 additional subspecies where more than one subspecies of a species exists in the Philippines). Four tables are provided: 1) a breakdown of the number of species and endemic species/subspecies for each subfamily, tribe and genus of Philippine Sphingidae; 2) an evaluation of the number of species as well as endemic species/subspecies per island for the nine largest islands of the Philippines plus one small island group for comparison; 3) an evaluation of the Sphingidae endemicity for each of Vane-Wright’s (1990) faunal regions. From these tables it can be readily deduced that the highest species counts can be encountered on the islands of Palawan (73 species), Luzon (72), Mindanao, Leyte and Negros (62 each). -

Redalyc.A Review of the Genus Ectropa Wallengren, 1863 with Descriptions of a New Genus and Six New Species (Lepidoptera: Chryso
SHILAP Revista de Lepidopterología ISSN: 0300-5267 [email protected] Sociedad Hispano-Luso-Americana de Lepidopterología España Kurshakov, P. A.; Zolotuhin, V. V. A review of the genus Ectropa Wallengren, 1863 with descriptions of a new genus and six new species (Lepidoptera: Chrysopolomidae) SHILAP Revista de Lepidopterología, vol. 41, núm. 164, octubre-diciembre, 2013, pp. 431-447 Sociedad Hispano-Luso-Americana de Lepidopterología Madrid, España Available in: http://www.redalyc.org/articulo.oa?id=45530406003 How to cite Complete issue Scientific Information System More information about this article Network of Scientific Journals from Latin America, the Caribbean, Spain and Portugal Journal's homepage in redalyc.org Non-profit academic project, developed under the open access initiative 431-447 A review of the genus 2/12/13 16:37 Página 431 SHILAP Revta. lepid., 41 (164), diciembre 2013: 431-447 eISSN: 2340-4078 ISSN: 0300-5267 A review of the genus Ectropa Wallengren, 1863 with descriptions of a new genus and six new species (Lepidoptera: Chrysopolomidae) P. A. Kurshakov & V. V. Zolotuhin Summary The genus Ectropa Wallengren, 1863, is revised, and a new species, E. adam Kurshakov & Zolotuhin, sp. n., is described. Based on the wing pattern and male genitalia characters, a new genus Ectropona Kurshakov & Zolotuhin, gen. n., related to Ectropa, is erected here, with Ectropona dargei Kurshakov & Zolotuhin, sp. n., as a type-species. Five more new species are also described: Ectropona dargei Kurshakov & Zolotuhin, sp. n., E. larsa Kurshakov & Zolotuhin, sp. n., E. kubwa Kurshakov & Zolotuhin, sp. n., E. aarviki Kurshakov & Zolotuhin, sp. n., and E. revelli Kurshakov & Zolotuhin, sp. -
Lepidoptera, Endromidae) in China
A peer-reviewed open-access journal ZooKeys 127: 29–42 (2011)The genusAndraca (Lepidoptera, Endromidae) in China... 29 doi: 10.3897/zookeys.127.928 RESEARCH artICLE www.zookeys.org Launched to accelerate biodiversity research The genus Andraca (Lepidoptera, Endromidae) in China with descriptions of a new species Xing Wang1,†, Ling Zeng2,‡, Min Wang2,§ 1 Department of Entomology, South China Agricultural University, Guangzhou, Guangdong 510640, P.R. China. Present address: Institute of Entomology, College of Biosafety Science and Technology, Hunan Agricul- tural University, Changsha 410128 Hunan, China; and Provincial Key Laboratory for Biology and Control of Plant Diseases and Insect Pests, Changsha 410128, Hunan, China 2 Department of Entomology, South China Agricultural University, Guangzhou, Guangdong 510640, P.R. China † urn:lsid:zoobank.org:author:F8727887-0014-42D4-BA68-21B3009E8C7F ‡ urn:lsid:zoobank.org:author:7981BF0E-D1F8-43CA-A505-72DBBA140023 § urn:lsid:zoobank.org:author:D683614E-1F58-4CA8-9D80-B23BD41947A2 Corresponding author: Ling Zeng ([email protected]) Academic editor: N. Wahlberg | Received 21 January 2011 | Accepted 15 August 2011 | Published 8 September 2011 urn:lsid:zoobank.org:pub:33D4BBFB-4B7D-4BBC-B34C-17D2E7F99F67 Citation: Wang X, Zeng L, Wang M (2011) The genus Andraca (Lepidoptera, Endromidae) in China with descriptions of a new species. ZooKeys 127: 29–42. doi: 10.3897/zookeys.127.928 Abstract The six species of the genus Andraca Walker hitherto known from China are reviewed, and a new species, A. gongshanensis, sp. n., described from Yunnan Province, China. Adults and male genitalia of all exam- ined species are illustrated, together with a distributional map. A key to all seven Chinese Andraca species is provided. -

BIO 313 ANIMAL ECOLOGY Corrected
NATIONAL OPEN UNIVERSITY OF NIGERIA SCHOOL OF SCIENCE AND TECHNOLOGY COURSE CODE: BIO 314 COURSE TITLE: ANIMAL ECOLOGY 1 BIO 314: ANIMAL ECOLOGY Team Writers: Dr O.A. Olajuyigbe Department of Biology Adeyemi Colledge of Education, P.M.B. 520, Ondo, Ondo State Nigeria. Miss F.C. Olakolu Nigerian Institute for Oceanography and Marine Research, No 3 Wilmot Point Road, Bar-beach Bus-stop, Victoria Island, Lagos, Nigeria. Mrs H.O. Omogoriola Nigerian Institute for Oceanography and Marine Research, No 3 Wilmot Point Road, Bar-beach Bus-stop, Victoria Island, Lagos, Nigeria. EDITOR: Mrs Ajetomobi School of Agricultural Sciences Lagos State Polytechnic Ikorodu, Lagos 2 BIO 313 COURSE GUIDE Introduction Animal Ecology (313) is a first semester course. It is a two credit unit elective course which all students offering Bachelor of Science (BSc) in Biology can take. Animal ecology is an important area of study for scientists. It is the study of animals and how they related to each other as well as their environment. It can also be defined as the scientific study of interactions that determine the distribution and abundance of organisms. Since this is a course in animal ecology, we will focus on animals, which we will define fairly generally as organisms that can move around during some stages of their life and that must feed on other organisms or their products. There are various forms of animal ecology. This includes: • Behavioral ecology, the study of the behavior of the animals with relation to their environment and others • Population ecology, the study of the effects on the population of these animals • Marine ecology is the scientific study of marine-life habitat, populations, and interactions among organisms and the surrounding environment including their abiotic (non-living physical and chemical factors that affect the ability of organisms to survive and reproduce) and biotic factors (living things or the materials that directly or indirectly affect an organism in its environment). -

Lepidoptera, Sphingidae, Smerinthinae)
Lucas Waldvogel Cardoso Contribuição de marcadores morfológicos e moleculares na elucidação da Sistemática de Ambulycini (Lepidoptera, Sphingidae, Smerinthinae) Contribution of morphological and molecular markers to the elucidation of the Ambulycini systematics (Lepidoptera, Sphingidae, Smerinthinae) São Paulo 2015 Exemplar corrigido O original encontra-se disponível no Instituto de Biociências da USP Lucas Waldvogel Cardoso Contribuição de marcadores morfológicos e moleculares na elucidação da Sistemática de Ambulycini (Lepidoptera, Sphingidae, Smerinthinae) Contribution of morphological and molecular markers to the elucidation of the Ambulycini systematics (Lepidoptera, Sphingidae, Smerinthinae) Dissertação apresentada ao Instituto de Biociências da Universidade de São Paulo, para a obtenção de Título de Mestre em Ciências Biológicas, na Área de Zoologia. Orientador: Prof. Dr. Marcelo Duarte da Silva São Paulo 2015 Ficha Catalográfica Waldvogel Cardoso, Lucas Contribuição de marcadores morfológicos e moleculares na elucidação da Sistemática de Ambulycini (Lepidoptera, Sphingidae, Smerinthinae) 187 + iv pp. Dissertação (Mestrado) - Instituto de Biociências da Universidade de São Paulo. Departamento de Zoologia. 1. Ambulycini 2. Filogenia 3. Filogeografia 4. Conservação I. Universidade de São Paulo. Instituto de Biociências. Departamento de Zoologia. Comissão Julgadora: ________________________ _______________________ Prof(a). Dr(a). Prof(a). Dr(a). ______________________ Prof(a). Dr.(a). Orientador(a) Dedicatória À minha família, a quem devo todas as minhas conquistas. Epígrafe ‘‘The crux of the problem is that if we don’t know what is out there or how widely species are distributed, how can we convince people about the reality and form of the biodiversity crisis?’’ Riddle et al. (2011). Agradecimentos Inicialmente, ao Prof. Dr. Marcelo Duarte, a quem devo muito pela paciência e compreensão. -

Supercritical Carbon Dioxide Extraction of Oil from Clanis Bilineata (Lepidoptera), an Edible Insect
African Journal of Biotechnology Vol. 11(20), pp. 4607-4610, 8 March, 2012 Available online at http://www.academicjournals.org/AJB DOI: 10.5897/AJB11.4102 ISSN 1684–5315 © 2012 Academic Journals Full Length Research Paper Supercritical carbon dioxide extraction of oil from Clanis bilineata (Lepidoptera), an edible insect Shengjun Wu School of Marine Science and Technology, HuaiHai Institute of Technology, 59 Cangwu Road, Xinpu 222005, China. E- mail: [email protected]. Tel: 86-051885895427. Fax: 86-051885895428. Accepted 16 February, 2012 Oil was extracted from the dry meat of Clanis bilineata (Lepidoptera) using supercritical carbon dioxide in a continuous flow extractor. The following optimum extraction conditions were investigated: temperature, 35°C; pressure, 25 MPa; supercritical CO2 flow rate, 20 L/min and time, 60 min. Under these extraction conditions, the oil extraction rate reached up to 97% (w/w). The level of unsaturated fatty acids in the extracted oil was 63.21% (w/w). In addition, the level of linolenic acid, a functional fatty acid, was as high as 37.61% (w/w) of the total fatty acids. Results of the present work indicate that C. bilineata (Lepidoptera) may be a promising oil resource for humans. Key words: Supercritical CO2, Clanis bilineata, fatty acids, oil. INTRODUCTION Many studies on a variety of edible insects which grow in dried CB meat, 0.53 mm; supercritical CO2 flow rate, 20 L/min; the wild indicate that these insects are potential sources pressure, 25 MPa; temperature, 35°C and time, 60 min. of oil for food and medicine (Chen, 1997; Raksakantong et al., 2010; Wu et al., 2000). -

! 2013 Elena Tartaglia ALL RIGHTS RESERVED
!"#$%&" '()*+",+-.+/(0+" 122"3456,7"3'7'38'9" HAWKMOTH – FLOWER INTERACTIONS IN THE URBAN LANDSCAPE: SPHINGIDAE ECOLOGY, WITH A FOCUS ON THE GENUS HEMARIS By ELENA S. TARTAGLIA A Dissertation submitted to the Graduate School-New Brunswick Rutgers, The State University of New Jersey in partial fulfillment of the requirements for the degree of Doctor of Philosophy Graduate Program in Ecology and Evolution written under the direction of Dr. Steven N. Handel and approved by ________________________________________! ________________________________________ ________________________________________ ________________________________________ New Brunswick, New Jersey May 2013 ABSTRACT OF THE DISSERTATION Hawkmoth-Flower Interactions in the Urban Landscape: Sphingidae Ecology, With a Focus on the Genus Hemaris by ELENA S. TARTAGLIA Dissertation Director: Steven N. Handel ! In this dissertation I examined the ecology of moths of the family Sphingidae in New Jersey and elucidated some previously unknown aspects of their behavior as floral visitors. In Chapter 2, I investigated differences in moth abundance and diversity between urban and suburban habitat types. Suburban sites have higher moth abundance and diversity than urban sites. I compared nighttime light intensities across all sites to correlate increased nighttime light intensity with moth abundance and diversity. Urban sites had significantly higher nighttime light intensity, a factor that has been shown to negatively affect the behavior of moths. I analyzed moths’ diets based on pollen grains swabbed from the moths’ bodies. These data were inconclusive due to insufficient sample sizes. In Chapter 3, I examined similar questions regarding diurnal Sphingidae of the genus Hemaris and found that suburban sites had higher moth abundances and diversities than urban sites. -

Acoustic Communication in the Nocturnal Lepidoptera
Chapter 6 Acoustic Communication in the Nocturnal Lepidoptera Michael D. Greenfield Abstract Pair formation in moths typically involves pheromones, but some pyra- loid and noctuoid species use sound in mating communication. The signals are generally ultrasound, broadcast by males, and function in courtship. Long-range advertisement songs also occur which exhibit high convergence with commu- nication in other acoustic species such as orthopterans and anurans. Tympanal hearing with sensitivity to ultrasound in the context of bat avoidance behavior is widespread in the Lepidoptera, and phylogenetic inference indicates that such perception preceded the evolution of song. This sequence suggests that male song originated via the sensory bias mechanism, but the trajectory by which ances- tral defensive behavior in females—negative responses to bat echolocation sig- nals—may have evolved toward positive responses to male song remains unclear. Analyses of various species offer some insight to this improbable transition, and to the general process by which signals may evolve via the sensory bias mechanism. 6.1 Introduction The acoustic world of Lepidoptera remained for humans largely unknown, and this for good reason: It takes place mostly in the middle- to high-ultrasound fre- quency range, well beyond our sensitivity range. Thus, the discovery and detailed study of acoustically communicating moths came about only with the use of electronic instruments sensitive to these sound frequencies. Such equipment was invented following the 1930s, and instruments that could be readily applied in the field were only available since the 1980s. But the application of such equipment M. D. Greenfield (*) Institut de recherche sur la biologie de l’insecte (IRBI), CNRS UMR 7261, Parc de Grandmont, Université François Rabelais de Tours, 37200 Tours, France e-mail: [email protected] B. -

Phylogeny and Biogeography of Hawkmoths (Lepidoptera: Sphingidae): Evidence from Five Nuclear Genes
Phylogeny and Biogeography of Hawkmoths (Lepidoptera: Sphingidae): Evidence from Five Nuclear Genes Akito Y. Kawahara1*, Andre A. Mignault1, Jerome C. Regier2, Ian J. Kitching3, Charles Mitter1 1 Department of Entomology, College Park, Maryland, United States of America, 2 Center for Biosystems Research, University of Maryland Biotechnology Institute, College Park, Maryland, United States of America, 3 Department of Entomology, The Natural History Museum, London, United Kingdom Abstract Background: The 1400 species of hawkmoths (Lepidoptera: Sphingidae) comprise one of most conspicuous and well- studied groups of insects, and provide model systems for diverse biological disciplines. However, a robust phylogenetic framework for the family is currently lacking. Morphology is unable to confidently determine relationships among most groups. As a major step toward understanding relationships of this model group, we have undertaken the first large-scale molecular phylogenetic analysis of hawkmoths representing all subfamilies, tribes and subtribes. Methodology/Principal Findings: The data set consisted of 131 sphingid species and 6793 bp of sequence from five protein-coding nuclear genes. Maximum likelihood and parsimony analyses provided strong support for more than two- thirds of all nodes, including strong signal for or against nearly all of the fifteen current subfamily, tribal and sub-tribal groupings. Monophyly was strongly supported for some of these, including Macroglossinae, Sphinginae, Acherontiini, Ambulycini, Philampelini, Choerocampina, and Hemarina. Other groupings proved para- or polyphyletic, and will need significant redefinition; these include Smerinthinae, Smerinthini, Sphingini, Sphingulini, Dilophonotini, Dilophonotina, Macroglossini, and Macroglossina. The basal divergence, strongly supported, is between Macroglossinae and Smerinthinae+Sphinginae. All genes contribute significantly to the signal from the combined data set, and there is little conflict between genes. -

Newsletter #259
COOLEMAN RIDGE PARK CARE GROUP Newsletter February 2015 Previous Meeting - Sunday 18th Afterburn Following the November prescribed burn at January GAS Arawang Kathner Street, the blackened hillside has A grey and muggy morning saw a good turn- begun to glow. These yellow flowers haven’t out of volunteers. We welcomed back our just been *Hypericum perforatum St John’s octogenerian cyclist mate Gősta, as cheerful Wort, nor * Hypochaeris radicata Flatweed. and willing as ever. He sets us such a great We’re delighted to report that Tricoryne example! elatior Yellow Rush Lily has had a great Led by Alan, a scout party of Rob, Graham season – here’s Malcolm Gill’s photo! and Gősta set out after woody weeds, heading north along the base trail. Their quarry sighted, the scouts clambered up the stony hillside and attacked. Photos exist to show the slaughter. The rest of the mob stayed near our usual site, above the wooden bridge. As ever, Rohan and Doug found *Rubus fruticosus Blackberry and *Phalaris aquatica aplenty. Arminel and Linda bagged seeding *Vicia sp. Vetch just above the drain, then joined the lads. Pat The Friday Weeders have counted 40 hunted Bracken rhizomes for propagation. reappearing species. They are now attacking Malcolm joined us at a convivial morning tea. exposed weeds. *Eragrostis curvula Tall He had cycled from Kathner Street, weeding African Lovegrass tussocks near the dam are as he went. already carrying new seed! Future programme Weed Watching The Committee has appointed Linda to NB – Remember! We meet in the document and identify weeds. Linda is our mornings during the warmer months! first port of call if we notice a problem or if Sunday 15th February we want to check whether we have a problem. -
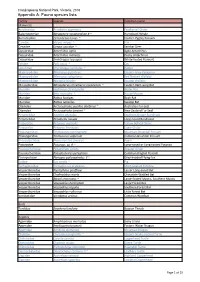
Report-VIC-Croajingolong National Park-Appendix A
Croajingolong National Park, Victoria, 2016 Appendix A: Fauna species lists Family Species Common name Mammals Acrobatidae Acrobates pygmaeus Feathertail Glider Balaenopteriae Megaptera novaeangliae # ~ Humpback Whale Burramyidae Cercartetus nanus ~ Eastern Pygmy Possum Canidae Vulpes vulpes ^ Fox Cervidae Cervus unicolor ^ Sambar Deer Dasyuridae Antechinus agilis Agile Antechinus Dasyuridae Antechinus mimetes Dusky Antechinus Dasyuridae Sminthopsis leucopus White-footed Dunnart Felidae Felis catus ^ Cat Leporidae Oryctolagus cuniculus ^ Rabbit Macropodidae Macropus giganteus Eastern Grey Kangaroo Macropodidae Macropus rufogriseus Red Necked Wallaby Macropodidae Wallabia bicolor Swamp Wallaby Miniopteridae Miniopterus schreibersii oceanensis ~ Eastern Bent-wing Bat Muridae Hydromys chrysogaster Water Rat Muridae Mus musculus ^ House Mouse Muridae Rattus fuscipes Bush Rat Muridae Rattus lutreolus Swamp Rat Otariidae Arctocephalus pusillus doriferus ~ Australian Fur-seal Otariidae Arctocephalus forsteri ~ New Zealand Fur Seal Peramelidae Isoodon obesulus Southern Brown Bandicoot Peramelidae Perameles nasuta Long-nosed Bandicoot Petauridae Petaurus australis Yellow Bellied Glider Petauridae Petaurus breviceps Sugar Glider Phalangeridae Trichosurus cunninghami Mountain Brushtail Possum Phalangeridae Trichosurus vulpecula Common Brushtail Possum Phascolarctidae Phascolarctos cinereus Koala Potoroidae Potorous sp. # ~ Long-nosed or Long-footed Potoroo Pseudocheiridae Petauroides volans Greater Glider Pseudocheiridae Pseudocheirus peregrinus