CHARACTERISATION of the HUMAN H^Atpase
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 T + is online at: average * The Journal of Immunology , 34 of which you can access for free at: 2016; 197:1477-1488; Prepublished online 1 July from submission to initial decision 4 weeks from acceptance to publication 2016; doi: 10.4049/jimmunol.1600589 http://www.jimmunol.org/content/197/4/1477 Molecular Profile of Tumor-Specific CD8 Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. Waugh, Sonia M. Leach, Brandon L. Moore, Tullia C. Bruno, Jonathan D. Buhrman and Jill E. Slansky J Immunol cites 95 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2016/07/01/jimmunol.160058 9.DCSupplemental This article http://www.jimmunol.org/content/197/4/1477.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 25, 2021. The Journal of Immunology Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. -

ATP6V0A2 Rabbit Polyclonal Antibody – TA308549 | Origene
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TA308549 ATP6V0A2 Rabbit Polyclonal Antibody Product data: Product Type: Primary Antibodies Applications: WB Recommended Dilution: WB:1:1000-1:10000 Reactivity: Human, Mouse Host: Rabbit Isotype: IgG Clonality: Polyclonal Immunogen: Recombinant fragment corresponding to a region within amino acids 156 and 434 of ATP6V0A2 (Uniprot ID#Q9Y487) Formulation: 1XPBS, 20% Glycerol (pH7). 0.01% Thimerosal was added as a preservative. Concentration: lot specific Purification: Purified by antigen-affinity chromatography. Conjugation: Unconjugated Storage: Store at -20°C as received. Stability: Stable for 12 months from date of receipt. Predicted Protein Size: 98 kDa Gene Name: ATPase H+ transporting V0 subunit a2 Database Link: NP_036595 Entrez Gene 21871 MouseEntrez Gene 23545 Human Q9Y487 Background: The multisubunit vacuolar-type proton pump (H(+)-ATPase or V-ATPase) is essential for acidification of diverse cellular components, including endosomes, lysosomes, clathrin-coated vesicles, secretory vesicles, and chromaffin granules, and it is found at high density in the plasma membrane of certain specialized cells. H(+)-ATPases are comprised of a peripheral V(1) domain and an integral membrane V(0) domain; ATP6V0A2 is a component of the V(0) domain (Smith et al., 2003 [PubMed 14580332]). [supplied by OMIM] Synonyms: A2; ARCL; ARCL2A; ATP6A2; -

Genomic and Functional Profiling of Duplicated Chromosome
Human Molecular Genetics, 2006, Vol. 15, No. 6 853–869 doi:10.1093/hmg/ddl004 Advance Access published on January 30, 2006 Genomic and functional profiling of duplicated chromosome 15 cell lines reveal regulatory alterations in UBE3A-associated ubiquitin–proteasome pathway processes Colin A. Baron1, Clifford G. Tepper2, Stephenie Y. Liu1, Ryan R. Davis1, Nicholas J. Wang3, N. Carolyn Schanen4 and Jeffrey P. Gregg1,* 1Department of Pathology and 2Department of Biochemistry and Molecular Medicine, University of California, Davis School of Medicine, Sacramento, CA 95817, USA, 3Department of Human Genetics, University of 4 California–Los Angeles, Los Angeles, CA, USA and Center for Pediatric Research, Nemours Biomedical Research, Downloaded from Alfred I. duPont Hospital for Children, Wilmington, DE, USA Received November 21, 2005; Revised and Accepted January 25, 2006 Autism is a complex neurodevelopmental disorder having both genetic and epigenetic etiological elements. hmg.oxfordjournals.org Isodicentric chromosome 15 (Idic15), characterized by duplications of the multi-disorder critical region of 15q11–q14, is a relatively common cytogenetic event. When the duplication involves maternally derived con- tent, this abnormality is strongly correlated with autism disorder. However, the mechanistic links between Idic15 and autism are ill-defined. To gain insight into the potential role of these duplications, we performed a comprehensive, genomics-based characterization of an in vitro model system consisting of lymphoblast cell lines derived from individuals with both autism and Idic15. Array-based comparative genomic hybridiz- ation using commercial single nucleotide polymorphism arrays was conducted and found to be capable of by guest on December 17, 2010 sub-classifying Idic15 samples by virtue of the lengths of the duplicated chromosomal region. -

The Role of the Golgi Apparatus in Disease (Review)
INTERNATIONAL JOURNAL OF MOleCular meDICine 47: 38, 2021 The role of the Golgi apparatus in disease (Review) JIANYANG LIU, YAN HUANG, TING LI, ZHENG JIANG, LIUWANG ZENG and ZHIPING HU Department of Neurology, Second Xiangya Hospital, Central South University, Changsha, Hunan 410011, P.R. China Received August 12, 2020; Accepted January 15, 2021 DOI: 10.3892/ijmm.2021.4871 Abstract. The Golgi apparatus is known to underpin many 1. Introduction important cellular homeostatic functions, including trafficking, sorting and modifications of proteins or lipids. These functions The Golgi apparatus is a processing and sorting hub in the are dysregulated in neurodegenerative diseases, cancer, infec‑ transport and targeting of soluble cargo proteins and lipids to tious diseases and cardiovascular diseases, and the number of different destinations in the cell (1). Considering its central disease‑related genes associated with Golgi apparatus is on the role in the secretory pathway, alterations in the structure increase. Recently, many studies have suggested that the muta‑ and function of the Golgi apparatus are expected to affect tions in the genes encoding Golgi resident proteins can trigger the homeostasis of cellular proteins and lipids. Increasing the occurrence of diseases. By summarizing the pathogenesis of evidence suggests that structural changes and functional these genetic diseases, it was found that most of these diseases disorder of the Golgi apparatus are involved in many human have defects in membrane trafficking. Such defects typically diseases such as neurodegenerative diseases (2‑4), ischemic result in mislocalization of proteins, impaired glycosylation of stroke (5,6), cardiovascular diseases (7,8), pulmonary arterial proteins, and the accumulation of undegraded proteins. -
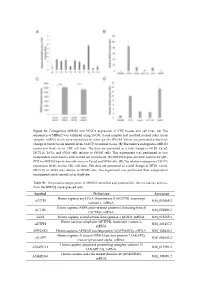
Figure S1. Endogenous MIR45
Figure S1. Endogenous MIR452 and VEGFA expression in CRC tissues and cell lines. (A) The expression of MIR452 was validated using 10 CRC tissue samples and matched normal colon tissue samples. miRNA levels were normalized to colon-specific RNU48. Values are presented as the fold- change in tumor tissue relative levels (ΔΔCT) to normal tissue. (B) The relative endogenous MIR452 expression levels in six CRC cell lines. The data are presented as a fold change in HT29, Caco2, HCT116, LoVo, and SW48 cells relative to SW480 cells. This experiment was performed as two independent experiments, each carried out in triplicate. (C) MIR452 expression level analysis by qRT- PCR for MIR452 transfection efficiency in Caco2 and SW48 cells. (D) The relative endogenous VEGFA expression levels in five CRC cell lines. The data are presented as a fold change in HT29, Caco2, HCT116, or SW48 cells relative to SW480 cells. This experiment was performed three independent experiments, each carried out in duplicate. Table S1. The putative target genes of MIR452 identified and predicted by the microarray analysis from the MIR452 overexpressed cells. Symbol Definition Accession Homo sapiens acyl-CoA thioesterase 8 (ACOT8), transcript ACOT8 NM_005469.2 variant 1, mRNA. Homo sapiens ARP6 actin-related protein 6 homolog (yeast) ACTR6 NM_022496.3 (ACTR6), mRNA. ADI1 Homo sapiens acireductone dioxygenase 1 (ADI1), mRNA. NM_018269.1 Homo sapiens aftiphilin (AFTPH), transcript variant 1, AFTPH NM_203437.2 mRNA. AHNAK2 Homo sapiens AHNAK nucleoprotein 2 (AHNAK2), mRNA. NM_138420.2 Homo sapiens A kinase (PRKA) anchor protein 7 (AKAP7), AKAP7 NM_004842.2 transcript variant alpha, mRNA. Homo sapiens anaphase promoting complex subunit 13 ANAPC13 NM_015391.2 (ANAPC13), mRNA. -

Autocrine IFN Signaling Inducing Profibrotic Fibroblast Responses By
Downloaded from http://www.jimmunol.org/ by guest on September 23, 2021 Inducing is online at: average * The Journal of Immunology , 11 of which you can access for free at: 2013; 191:2956-2966; Prepublished online 16 from submission to initial decision 4 weeks from acceptance to publication August 2013; doi: 10.4049/jimmunol.1300376 http://www.jimmunol.org/content/191/6/2956 A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Autocrine IFN Signaling Feng Fang, Kohtaro Ooka, Xiaoyong Sun, Ruchi Shah, Swati Bhattacharyya, Jun Wei and John Varga J Immunol cites 49 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130037 6.DC1 This article http://www.jimmunol.org/content/191/6/2956.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 23, 2021. The Journal of Immunology A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Inducing Autocrine IFN Signaling Feng Fang,* Kohtaro Ooka,* Xiaoyong Sun,† Ruchi Shah,* Swati Bhattacharyya,* Jun Wei,* and John Varga* Activation of TLR3 by exogenous microbial ligands or endogenous injury-associated ligands leads to production of type I IFN. -

Supplemental Figures 04 12 2017
Jung et al. 1 SUPPLEMENTAL FIGURES 2 3 Supplemental Figure 1. Clinical relevance of natural product methyltransferases (NPMTs) in brain disorders. (A) 4 Table summarizing characteristics of 11 NPMTs using data derived from the TCGA GBM and Rembrandt datasets for 5 relative expression levels and survival. In addition, published studies of the 11 NPMTs are summarized. (B) The 1 Jung et al. 6 expression levels of 10 NPMTs in glioblastoma versus non‐tumor brain are displayed in a heatmap, ranked by 7 significance and expression levels. *, p<0.05; **, p<0.01; ***, p<0.001. 8 2 Jung et al. 9 10 Supplemental Figure 2. Anatomical distribution of methyltransferase and metabolic signatures within 11 glioblastomas. The Ivy GAP dataset was downloaded and interrogated by histological structure for NNMT, NAMPT, 12 DNMT mRNA expression and selected gene expression signatures. The results are displayed on a heatmap. The 13 sample size of each histological region as indicated on the figure. 14 3 Jung et al. 15 16 Supplemental Figure 3. Altered expression of nicotinamide and nicotinate metabolism‐related enzymes in 17 glioblastoma. (A) Heatmap (fold change of expression) of whole 25 enzymes in the KEGG nicotinate and 18 nicotinamide metabolism gene set were analyzed in indicated glioblastoma expression datasets with Oncomine. 4 Jung et al. 19 Color bar intensity indicates percentile of fold change in glioblastoma relative to normal brain. (B) Nicotinamide and 20 nicotinate and methionine salvage pathways are displayed with the relative expression levels in glioblastoma 21 specimens in the TCGA GBM dataset indicated. 22 5 Jung et al. 23 24 Supplementary Figure 4. -

Table S1. 103 Ferroptosis-Related Genes Retrieved from the Genecards
Table S1. 103 ferroptosis-related genes retrieved from the GeneCards. Gene Symbol Description Category GPX4 Glutathione Peroxidase 4 Protein Coding AIFM2 Apoptosis Inducing Factor Mitochondria Associated 2 Protein Coding TP53 Tumor Protein P53 Protein Coding ACSL4 Acyl-CoA Synthetase Long Chain Family Member 4 Protein Coding SLC7A11 Solute Carrier Family 7 Member 11 Protein Coding VDAC2 Voltage Dependent Anion Channel 2 Protein Coding VDAC3 Voltage Dependent Anion Channel 3 Protein Coding ATG5 Autophagy Related 5 Protein Coding ATG7 Autophagy Related 7 Protein Coding NCOA4 Nuclear Receptor Coactivator 4 Protein Coding HMOX1 Heme Oxygenase 1 Protein Coding SLC3A2 Solute Carrier Family 3 Member 2 Protein Coding ALOX15 Arachidonate 15-Lipoxygenase Protein Coding BECN1 Beclin 1 Protein Coding PRKAA1 Protein Kinase AMP-Activated Catalytic Subunit Alpha 1 Protein Coding SAT1 Spermidine/Spermine N1-Acetyltransferase 1 Protein Coding NF2 Neurofibromin 2 Protein Coding YAP1 Yes1 Associated Transcriptional Regulator Protein Coding FTH1 Ferritin Heavy Chain 1 Protein Coding TF Transferrin Protein Coding TFRC Transferrin Receptor Protein Coding FTL Ferritin Light Chain Protein Coding CYBB Cytochrome B-245 Beta Chain Protein Coding GSS Glutathione Synthetase Protein Coding CP Ceruloplasmin Protein Coding PRNP Prion Protein Protein Coding SLC11A2 Solute Carrier Family 11 Member 2 Protein Coding SLC40A1 Solute Carrier Family 40 Member 1 Protein Coding STEAP3 STEAP3 Metalloreductase Protein Coding ACSL1 Acyl-CoA Synthetase Long Chain Family Member 1 Protein -

ATP6V0A2 Gene Atpase H+ Transporting V0 Subunit A2
ATP6V0A2 gene ATPase H+ transporting V0 subunit a2 Normal Function The ATP6V0A2 gene provides instructions for making one part, the a2 subunit, of a large protein complex (a group of proteins that work together). This protein complex is known as a vacuolar H+-ATPase (V-ATPase). A V-ATPase acts as a pump to move positively charged hydrogen atoms (protons) across cell membranes. V-ATPases are embedded in the membranes surrounding cells, where they transport protons into and out of cells. This movement of protons helps regulate the relative acidity (pH) of cells and their surrounding environment. Tight control of pH is necessary for most biological reactions to proceed properly. Within cells, V-ATPases help regulate the pH of particular cell compartments. These compartments include endosomes and lysosomes, which digest and recycle materials that the cell no longer needs. Studies suggest that V-ATPases are also involved in the movement (trafficking) of small sac-like structures called vesicles. Vesicles transport many types of molecules within cells. V-ATPases also play a key role in a complex process called glycosylation, in which proteins are modified by adding sugar molecules. Glycosylation is necessary for the normal function of many different kinds of proteins. V-ATPases regulate the pH of a cellular structure called the Golgi apparatus, where glycosylation occurs. Health Conditions Related to Genetic Changes Cutis laxa More than 40 variants (also known as mutations) in the ATP6V0A2 gene have been identified in people with cutis laxa. ATP6V0A2 variants cause a form of the disorder called autosomal recessive cutis laxa type 2A (ARCL2A), which is characterized by loose, sagging skin; distinctive facial features; and larger than normal spaces ( fontanelles) between the skull bones that close later than usual. -

Sensors and Regulators of Intracellular Ph
REVIEWS Sensors and regulators of intracellular pH Joseph R. Casey*, Sergio Grinstein‡ and John Orlowski§ Abstract | Protons dictate the charge and structure of macromolecules and are used as energy currency by eukaryotic cells. The unique function of individual organelles therefore depends on the establishment and stringent maintenance of a distinct pH. This, in turn, requires a means to sense the prevailing pH and to respond to deviations from the norm with effective mechanisms to transport, produce or consume proton equivalents. A dynamic, finely tuned balance between proton-extruding and proton-importing processes underlies pH homeostasis not only in the cytosol, but in other cellular compartments as well. Proton-motive force Eukaryotic cells are highly compartmentalized, seg- As a result, βintrinsic is comparatively low at physiological regating specific functions within membrane-bound pH (10–20 mM at pH 7.2), increasing at more extreme (ψH+). The driving force for proton (or equivalent) organelles. Compartmentalization probably evolved from values (for example, 40 mM at pH 6.4)3. This apparent movement, consisting of the the need to provide distinct environmental conditions shortcoming is alleviated by the second component of the proton concentration gradient for the optimal operation of individual metabolic pathways, total buffering capacity, β . Mammalian cells are con- and the transmembrane HCO3– electrical potential. and also to store energy as electrochemical gradients tinuously exposed to CO2, which is uncharged and readily across the membrane dielectric. Protons (and proton traverses most biological membranes4. The hydration of pH buffering capacity equivalents) have a crucial role in this context. Virtually CO2 and the subsequent deprotonation of carbonic acid A measure of the ability of a all proteins depend on pH to maintain their structure generate HCO –, an effective proton buffer (BOX 1). -
Pulmonary Hypertension Remodels the Genomic Fabrics of Major Functional Pathways
G C A T T A C G G C A T genes Article Pulmonary Hypertension Remodels the Genomic Fabrics of Major Functional Pathways Rajamma Mathew 1,2, Jing Huang 1, Sanda Iacobas 3 and Dumitru A. Iacobas 4,* 1 Department of Pediatrics, New York Medical College, Valhalla, NY 10595, USA; [email protected] (R.M.); [email protected] (J.H.) 2 Department of Physiology, New York Medical College, Valhalla, NY 10595, USA 3 Department of Pathology, New York Medical College, Valhalla, NY 10595, USA; [email protected] 4 Personalized Genomics Laboratory, Center for Computational Systems Biology, Roy G Perry College of Engineering, Prairie View A&M University, Prairie View, TX 77446, USA * Correspondence: [email protected]; Tel.: +1-936-261-9926 Received: 17 December 2019; Accepted: 21 January 2020; Published: 23 January 2020 Abstract: Pulmonary hypertension (PH) is a serious disorder with high morbidity and mortality rate. We analyzed the right-ventricular systolic pressure (RVSP), right-ventricular hypertrophy (RVH), lung histology, and transcriptomes of six-week-old male rats with PH induced by (1) hypoxia (HO), (2) administration of monocrotaline (CM), or (3) administration of monocrotaline and exposure to hypoxia (HM). The results in PH rats were compared to those in control rats (CO). After four weeks exposure, increased RVSP and RVH, pulmonary arterial wall thickening, and alteration of the lung transcriptome were observed in all PH groups. The HM group exhibited the largest alterations, as well as neointimal lesions and obliteration of the lumen in small arteries. We found that PH increased the expression of caveolin1, matrix metallopeptidase 2, and numerous inflammatory and cell proliferation genes. -

D-Serine, an Emerging Biomarker of Kidney Diseases, Is a Hidden Substrate of Sodium-Coupled Monocarboxylate Transporters
bioRxiv preprint doi: https://doi.org/10.1101/2020.08.10.244822; this version posted August 10, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Wiriyasermkul, et al. 1 D-Serine, an emerging biomarker of kidney diseases, is a hidden substrate of 2 sodium-coupled monocarboxylate transporters 3 4 Pattama Wiriyasermkul1, Satomi Moriyama1, Yoko Tanaka1, Pornparn Kongpracha1, Nodoka 5 Nakamae1, Masataka Suzuki2, Tomonori Kimura3,4, Masashi Mita5, Jumpei Sasabe2, Shushi 6 Nagamori1,# 7 8 1Department of Collaborative Research for Biomolecular Dynamics, Nara Medical 9 University, Nara, Japan 10 2Department of Pharmacology, Keio University School of Medicine, Tokyo, Japan 11 3KAGAMI Project, 4Reverse Translational Research Project, Center for Rare Disease 12 Research, National Institutes of Biomedical Innovation, Health and Nutrition (NIBIOHN). 13 Osaka, Japan 14 5KAGAMI Inc., Osaka, Japan 15 16 # Corresponding contact information: [email protected] 17 18 19 20 21 Keywords: transporter, proteome, ischemia-reperfusion injury (IRI), acute kidney injury 22 (AKI), chronic kidney diseases (CKD), membrane 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.08.10.244822; this version posted August 10, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Wiriyasermkul, et al. 23 Abstract 24 Delay in diagnosis of renal injury has profound impacts on morbidity and mortality.