ATP6V0A2 Gene Atpase H+ Transporting V0 Subunit A2
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 T + is online at: average * The Journal of Immunology , 34 of which you can access for free at: 2016; 197:1477-1488; Prepublished online 1 July from submission to initial decision 4 weeks from acceptance to publication 2016; doi: 10.4049/jimmunol.1600589 http://www.jimmunol.org/content/197/4/1477 Molecular Profile of Tumor-Specific CD8 Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. Waugh, Sonia M. Leach, Brandon L. Moore, Tullia C. Bruno, Jonathan D. Buhrman and Jill E. Slansky J Immunol cites 95 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2016/07/01/jimmunol.160058 9.DCSupplemental This article http://www.jimmunol.org/content/197/4/1477.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 25, 2021. The Journal of Immunology Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. -

ATP6V0A2 Rabbit Polyclonal Antibody – TA308549 | Origene
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TA308549 ATP6V0A2 Rabbit Polyclonal Antibody Product data: Product Type: Primary Antibodies Applications: WB Recommended Dilution: WB:1:1000-1:10000 Reactivity: Human, Mouse Host: Rabbit Isotype: IgG Clonality: Polyclonal Immunogen: Recombinant fragment corresponding to a region within amino acids 156 and 434 of ATP6V0A2 (Uniprot ID#Q9Y487) Formulation: 1XPBS, 20% Glycerol (pH7). 0.01% Thimerosal was added as a preservative. Concentration: lot specific Purification: Purified by antigen-affinity chromatography. Conjugation: Unconjugated Storage: Store at -20°C as received. Stability: Stable for 12 months from date of receipt. Predicted Protein Size: 98 kDa Gene Name: ATPase H+ transporting V0 subunit a2 Database Link: NP_036595 Entrez Gene 21871 MouseEntrez Gene 23545 Human Q9Y487 Background: The multisubunit vacuolar-type proton pump (H(+)-ATPase or V-ATPase) is essential for acidification of diverse cellular components, including endosomes, lysosomes, clathrin-coated vesicles, secretory vesicles, and chromaffin granules, and it is found at high density in the plasma membrane of certain specialized cells. H(+)-ATPases are comprised of a peripheral V(1) domain and an integral membrane V(0) domain; ATP6V0A2 is a component of the V(0) domain (Smith et al., 2003 [PubMed 14580332]). [supplied by OMIM] Synonyms: A2; ARCL; ARCL2A; ATP6A2; -

Genomic and Functional Profiling of Duplicated Chromosome
Human Molecular Genetics, 2006, Vol. 15, No. 6 853–869 doi:10.1093/hmg/ddl004 Advance Access published on January 30, 2006 Genomic and functional profiling of duplicated chromosome 15 cell lines reveal regulatory alterations in UBE3A-associated ubiquitin–proteasome pathway processes Colin A. Baron1, Clifford G. Tepper2, Stephenie Y. Liu1, Ryan R. Davis1, Nicholas J. Wang3, N. Carolyn Schanen4 and Jeffrey P. Gregg1,* 1Department of Pathology and 2Department of Biochemistry and Molecular Medicine, University of California, Davis School of Medicine, Sacramento, CA 95817, USA, 3Department of Human Genetics, University of 4 California–Los Angeles, Los Angeles, CA, USA and Center for Pediatric Research, Nemours Biomedical Research, Downloaded from Alfred I. duPont Hospital for Children, Wilmington, DE, USA Received November 21, 2005; Revised and Accepted January 25, 2006 Autism is a complex neurodevelopmental disorder having both genetic and epigenetic etiological elements. hmg.oxfordjournals.org Isodicentric chromosome 15 (Idic15), characterized by duplications of the multi-disorder critical region of 15q11–q14, is a relatively common cytogenetic event. When the duplication involves maternally derived con- tent, this abnormality is strongly correlated with autism disorder. However, the mechanistic links between Idic15 and autism are ill-defined. To gain insight into the potential role of these duplications, we performed a comprehensive, genomics-based characterization of an in vitro model system consisting of lymphoblast cell lines derived from individuals with both autism and Idic15. Array-based comparative genomic hybridiz- ation using commercial single nucleotide polymorphism arrays was conducted and found to be capable of by guest on December 17, 2010 sub-classifying Idic15 samples by virtue of the lengths of the duplicated chromosomal region. -

WO 2015/048577 A2 April 2015 (02.04.2015) W P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2015/048577 A2 April 2015 (02.04.2015) W P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every A61K 48/00 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (21) International Application Number: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, PCT/US20 14/057905 DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, 26 September 2014 (26.09.2014) KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (25) Filing Language: English PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, (26) Publication Language: English SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: 61/883,925 27 September 2013 (27.09.2013) US (84) Designated States (unless otherwise indicated, for every 61/898,043 31 October 2013 (3 1. 10.2013) US kind of regional protection available): ARIPO (BW, GH, GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, (71) Applicant: EDITAS MEDICINE, INC. -

Metabolic Cutis Laxa Syndromes
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Springer - Publisher Connector J Inherit Metab Dis (2011) 34:907–916 DOI 10.1007/s10545-011-9305-9 CDG - AN UPDATE Metabolic cutis laxa syndromes Miski Mohamed & Dorus Kouwenberg & Thatjana Gardeitchik & Uwe Kornak & Ron A. Wevers & Eva Morava Received: 28 October 2010 /Revised: 11 February 2011 /Accepted: 17 February 2011 /Published online: 23 March 2011 # The Author(s) 2011. This article is published with open access at Springerlink.com Abstract Cutis laxa is a rare skin disorder characterized by Disorders of Glycosylation (CDG). Since then several wrinkled, redundant, inelastic and sagging skin due to inborn errors of metabolism with cutis laxa have been defective synthesis of elastic fibers and other proteins of the described with variable severity. These include P5CS, extracellular matrix. Wrinkled, inelastic skin occurs in ATP6V0A2-CDG and PYCR1 defects. In spite of the many cases as an acquired condition. Syndromic forms of evolving number of cutis laxa-related diseases a large part cutis laxa, however, are caused by diverse genetic defects, of the cases remain genetically unsolved. In metabolic cutis mostly coding for structural extracellular matrix proteins. laxa syndromes the clinical and laboratory features might Surprisingly a number of metabolic disorders have been partially overlap, however there are some distinct, discrim- also found to be associated with inherited cutis laxa. inative features. In this review on metabolic diseases Menkes disease was the first metabolic disease reported causing cutis laxa we offer a practical approach for the with old-looking, wrinkled skin. -

Discriminative Features in Three Autosomal Recessive Cutis Laxa Syndromes: Cutis Laxa IIA, Cutis Laxa IIB, and Geroderma Osteoplastica
International Journal of Molecular Sciences Review Discriminative Features in Three Autosomal Recessive Cutis Laxa Syndromes: Cutis Laxa IIA, Cutis Laxa IIB, and Geroderma Osteoplastica Ariana Kariminejad 1,*, Fariba Afroozan 1, Bita Bozorgmehr 1, Alireza Ghanadan 2,3, Susan Akbaroghli 4, Hamid Reza Khorram Khorshid 5, Faezeh Mojahedi 6, Aria Setoodeh 7, Abigail Loh 8, Yu Xuan Tan 8, Nathalie Escande-Beillard 8, Fransiska Malfait 9, Bruno Reversade 8, Thatjana Gardeitchik 10 and Eva Morava 10,11 1 Kariminejad-Najmabadi Pathology & Genetics Center, #2, 4th Street, Hasan Seyf Street, Sanat Square, Tehran 14667-13713, Iran; [email protected] (F.A.); [email protected] (B.B.) 2 Department of Dermatopathology, Razi Dermatology Hospital, Tehran University of Medical Sciences, Tehran 14167-53955, Iran; [email protected] 3 Department of Pathology, Cancer Institute, Imam Khomeini Hospital Complex, Tehran University of Medical Sciences, Tehran 14197-33141, Iran 4 Clinical Genetics Division, Mofid Children’s Hospital, Faculty of Medicine, Shahid Beheshti University of Medical Sciences, Tehran 15514-15468, Iran; [email protected] 5 Genetic Research Centre, University of Social Welfare and Rehabilitation Sciences, Tehran 19857-13834, Iran; [email protected] 6 Mashhad Medical Genetic Counseling Center, Social Welfare and Rehabilitation Organization, Mashhad 91767-61999, Iran; [email protected] 7 Division of Pediatric Endocrinology and Inherited Metabolic Disorders, Department of Pediatrics, Tehran University of Medical Sciences, Tehran -

The Role of the Golgi Apparatus in Disease (Review)
INTERNATIONAL JOURNAL OF MOleCular meDICine 47: 38, 2021 The role of the Golgi apparatus in disease (Review) JIANYANG LIU, YAN HUANG, TING LI, ZHENG JIANG, LIUWANG ZENG and ZHIPING HU Department of Neurology, Second Xiangya Hospital, Central South University, Changsha, Hunan 410011, P.R. China Received August 12, 2020; Accepted January 15, 2021 DOI: 10.3892/ijmm.2021.4871 Abstract. The Golgi apparatus is known to underpin many 1. Introduction important cellular homeostatic functions, including trafficking, sorting and modifications of proteins or lipids. These functions The Golgi apparatus is a processing and sorting hub in the are dysregulated in neurodegenerative diseases, cancer, infec‑ transport and targeting of soluble cargo proteins and lipids to tious diseases and cardiovascular diseases, and the number of different destinations in the cell (1). Considering its central disease‑related genes associated with Golgi apparatus is on the role in the secretory pathway, alterations in the structure increase. Recently, many studies have suggested that the muta‑ and function of the Golgi apparatus are expected to affect tions in the genes encoding Golgi resident proteins can trigger the homeostasis of cellular proteins and lipids. Increasing the occurrence of diseases. By summarizing the pathogenesis of evidence suggests that structural changes and functional these genetic diseases, it was found that most of these diseases disorder of the Golgi apparatus are involved in many human have defects in membrane trafficking. Such defects typically diseases such as neurodegenerative diseases (2‑4), ischemic result in mislocalization of proteins, impaired glycosylation of stroke (5,6), cardiovascular diseases (7,8), pulmonary arterial proteins, and the accumulation of undegraded proteins. -
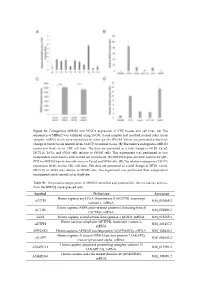
Figure S1. Endogenous MIR45
Figure S1. Endogenous MIR452 and VEGFA expression in CRC tissues and cell lines. (A) The expression of MIR452 was validated using 10 CRC tissue samples and matched normal colon tissue samples. miRNA levels were normalized to colon-specific RNU48. Values are presented as the fold- change in tumor tissue relative levels (ΔΔCT) to normal tissue. (B) The relative endogenous MIR452 expression levels in six CRC cell lines. The data are presented as a fold change in HT29, Caco2, HCT116, LoVo, and SW48 cells relative to SW480 cells. This experiment was performed as two independent experiments, each carried out in triplicate. (C) MIR452 expression level analysis by qRT- PCR for MIR452 transfection efficiency in Caco2 and SW48 cells. (D) The relative endogenous VEGFA expression levels in five CRC cell lines. The data are presented as a fold change in HT29, Caco2, HCT116, or SW48 cells relative to SW480 cells. This experiment was performed three independent experiments, each carried out in duplicate. Table S1. The putative target genes of MIR452 identified and predicted by the microarray analysis from the MIR452 overexpressed cells. Symbol Definition Accession Homo sapiens acyl-CoA thioesterase 8 (ACOT8), transcript ACOT8 NM_005469.2 variant 1, mRNA. Homo sapiens ARP6 actin-related protein 6 homolog (yeast) ACTR6 NM_022496.3 (ACTR6), mRNA. ADI1 Homo sapiens acireductone dioxygenase 1 (ADI1), mRNA. NM_018269.1 Homo sapiens aftiphilin (AFTPH), transcript variant 1, AFTPH NM_203437.2 mRNA. AHNAK2 Homo sapiens AHNAK nucleoprotein 2 (AHNAK2), mRNA. NM_138420.2 Homo sapiens A kinase (PRKA) anchor protein 7 (AKAP7), AKAP7 NM_004842.2 transcript variant alpha, mRNA. Homo sapiens anaphase promoting complex subunit 13 ANAPC13 NM_015391.2 (ANAPC13), mRNA. -

Autocrine IFN Signaling Inducing Profibrotic Fibroblast Responses By
Downloaded from http://www.jimmunol.org/ by guest on September 23, 2021 Inducing is online at: average * The Journal of Immunology , 11 of which you can access for free at: 2013; 191:2956-2966; Prepublished online 16 from submission to initial decision 4 weeks from acceptance to publication August 2013; doi: 10.4049/jimmunol.1300376 http://www.jimmunol.org/content/191/6/2956 A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Autocrine IFN Signaling Feng Fang, Kohtaro Ooka, Xiaoyong Sun, Ruchi Shah, Swati Bhattacharyya, Jun Wei and John Varga J Immunol cites 49 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130037 6.DC1 This article http://www.jimmunol.org/content/191/6/2956.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 23, 2021. The Journal of Immunology A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Inducing Autocrine IFN Signaling Feng Fang,* Kohtaro Ooka,* Xiaoyong Sun,† Ruchi Shah,* Swati Bhattacharyya,* Jun Wei,* and John Varga* Activation of TLR3 by exogenous microbial ligands or endogenous injury-associated ligands leads to production of type I IFN. -

Supplemental Figures 04 12 2017
Jung et al. 1 SUPPLEMENTAL FIGURES 2 3 Supplemental Figure 1. Clinical relevance of natural product methyltransferases (NPMTs) in brain disorders. (A) 4 Table summarizing characteristics of 11 NPMTs using data derived from the TCGA GBM and Rembrandt datasets for 5 relative expression levels and survival. In addition, published studies of the 11 NPMTs are summarized. (B) The 1 Jung et al. 6 expression levels of 10 NPMTs in glioblastoma versus non‐tumor brain are displayed in a heatmap, ranked by 7 significance and expression levels. *, p<0.05; **, p<0.01; ***, p<0.001. 8 2 Jung et al. 9 10 Supplemental Figure 2. Anatomical distribution of methyltransferase and metabolic signatures within 11 glioblastomas. The Ivy GAP dataset was downloaded and interrogated by histological structure for NNMT, NAMPT, 12 DNMT mRNA expression and selected gene expression signatures. The results are displayed on a heatmap. The 13 sample size of each histological region as indicated on the figure. 14 3 Jung et al. 15 16 Supplemental Figure 3. Altered expression of nicotinamide and nicotinate metabolism‐related enzymes in 17 glioblastoma. (A) Heatmap (fold change of expression) of whole 25 enzymes in the KEGG nicotinate and 18 nicotinamide metabolism gene set were analyzed in indicated glioblastoma expression datasets with Oncomine. 4 Jung et al. 19 Color bar intensity indicates percentile of fold change in glioblastoma relative to normal brain. (B) Nicotinamide and 20 nicotinate and methionine salvage pathways are displayed with the relative expression levels in glioblastoma 21 specimens in the TCGA GBM dataset indicated. 22 5 Jung et al. 23 24 Supplementary Figure 4. -

Blueprint Genetics Congenital Disorders of Glycosylation Panel
Congenital Disorders of Glycosylation Panel Test code: ME1901 Is a 48 gene panel that includes assessment of non-coding variants. Is ideal for patients with a clinical suspicion of a congenital disorder of N-linked glycosylation or combined defects of glycosylation affecting both the N-linked and O-linked glycosylation pathways. The genes on this panel are included in the Comprehensive Metabolism Panel. About Congenital Disorders of Glycosylation Most subtypes of congenital disorders of glycosylation (CDG) are classified as disorders of N-glycosylation, which involves carbohydrates called N-linked oligosaccharides. These oligosaccharides are created in a specific order to create specific sugar trees, which are then attached to proteins on various cells. Disorders of N-glycosylation are due to an enzyme deficiency or other malfunction somewhere along the N-glycosylation pathway. There are 42 different enzymes in the pathway; any of them may be mutated and cause a disorder belonging to this group. Different mutated enzymes cause different phenotypes. Congenital disorders of N-linked glycosylation are a genetically and phenotypically heterogeneous group of diseases. Most commonly, symptoms begin in early infancy. Manifestations range from mild to severe, involving only protein-losing enteropathy and hypoglycemia or severe intellectual disability with malfunction of several organs. Sometimes the disorder may be fatal. Most patients require nutritional supplements. Most of the individual disorders have been observed only in a very limited number of patients. The most common ones are PMM2-related disorder (approximately 700 patients reported), MPI-related disorder (>20 patients) and ALG6-related disorder (>30 patients). Other types of disorder are extremely rare. -

Global Journal of Medical Research: K I Nterdisciplinary
Online ISSN : 2249-4618 Print ISSN : 0975-5888 DOI : 10.17406/GJMRA Bafqs Miners Burnout Elevated Nutritional Needs Thérapeutiques Et Pronostiques Professional Community Training VOLUME 16 ISSUE 4 VERSION 1.0 Global Journal of Medical Research: k Interdisciplinary Global Journal of Medical Research: k Interdisciplinary Volume 16 Issue 4 (Ver. 1.0) Open Association of Research Society © Global Journal of Medical Global Journals Inc. Research . 2016. (A Delaware USA Incorporation with “Good Standing”; Reg. Number: 0423089) Sponsors:Open Association of Research Society All rights reserved. Open Scientific Standards This is a special issue published in version 1.0 Publisher’s Headquarters office of “Global Journal of Medical Research.” By Global Journals Inc. Global Journals ® Headquarters All articles are open access articles distributed 945th Concord Streets, under “Global Journal of Medical Research” Framingham Massachusetts Pin: 01701, Reading License, which permits restricted use. United States of America Entire contents are copyright by of “Global USA Toll Free: +001-888-839-7392 Journal of Medical Research” unless otherwise noted on specific articles. USA Toll Free Fax: +001-888-839-7392 No part of this publication may be reproduced Offset Typesetting or transmitted in any form or by any means, electronic or mechanical, including Global Journals Incorporated photocopy, recording, or any information storage and retrieval system, without written 2nd, Lansdowne, Lansdowne Rd., Croydon-Surrey, permission. Pin: CR9 2ER, United Kingdom The opinions and statements made in this book are those of the authors concerned. Packaging & Continental Dispatching Ultraculture has not verified and neither confirms nor denies any of the foregoing and Global Journals no warranty or fitness is implied.