Sexually Transmitted Diseases
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Official Nh Dhhs Health Alert
THIS IS AN OFFICIAL NH DHHS HEALTH ALERT Distributed by the NH Health Alert Network [email protected] May 18, 2018, 1300 EDT (1:00 PM EDT) NH-HAN 20180518 Tickborne Diseases in New Hampshire Key Points and Recommendations: 1. Blacklegged ticks transmit at least five different infections in New Hampshire (NH): Lyme disease, Anaplasma, Babesia, Powassan virus, and Borrelia miyamotoi. 2. NH has one of the highest rates of Lyme disease in the nation, and 50-60% of blacklegged ticks sampled from across NH have been found to be infected with Borrelia burgdorferi, the bacterium that causes Lyme disease. 3. NH has experienced a significant increase in human cases of anaplasmosis, with cases more than doubling from 2016 to 2017. The reason for the increase is unknown at this time. 4. The number of new cases of babesiosis also increased in 2017; because Babesia can be transmitted through blood transfusions in addition to tick bites, providers should ask patients with suspected babesiosis whether they have donated blood or received a blood transfusion. 5. Powassan is a newer tickborne disease which has been identified in three NH residents during past seasons in 2013, 2016 and 2017. While uncommon, Powassan can cause a debilitating neurological illness, so providers should maintain an index of suspicion for patients presenting with an unexplained meningoencephalitis. 6. Borrelia miyamotoi infection usually presents with a nonspecific febrile illness similar to other tickborne diseases like anaplasmosis, and has recently been identified in one NH resident. Tests for Lyme disease do not reliably detect Borrelia miyamotoi, so providers should consider specific testing for Borrelia miyamotoi (see Attachment 1) and other pathogens if testing for Lyme disease is negative but a tickborne disease is still suspected. -

Are You Suprised ?
B DAMB 721 Microbiology Final Exam B 100 points December 11, 2006 Your name (Print Clearly): _____________________________________________ I. Matching: The questions below consist of headings followed by a list of phrases. For each phrase select the heading that best describes that phrase. The headings may be used once, more than once or not at all. Mark the answer in Part 2 of your answer sheet. 1. capsid 7. CD4 2. Chlamydia pneumoniae 8. Enterococcus faecalis 3. oncogenic 9. hyaluronidase 4. pyruvate 10. interferon 5. Koplik’s spot 11. hydrophilic viruses 6. congenital Treponema pallidum 12. Streptococcus pyogenes 1. “spreading factor” produced by members of the staphylococci, streptococci and clostridia 2. viral protein coat 3. central intermediate in bacterial fermentation 4. persistant endodontic infections 5. a cause of atypical pneumonia 6. nonspecific defense against viral infection 7. has a rudimentary life cycle 8. HIV receptor 9. Hutchinson’s Triad 10. measles 11. resistant to disinfection 12. β-hemolytic, bacitracin sensitive, cause of suppurative pharyngitis 2 Matching (Continued): The questions below consist of diseases followed by a list of etiologic agents. Match each disease with the etiologic agent. Continue using Part 2 of your answer sheet. 1. dysentery 6. Legionnaire’s 2. botulism 7. gas gangrene 3. cholera 8. tuberculosis 4. diphtheria 9. necrotizing fascitis 5. enteric fever 10. pneumoniae/meningitis 13. Clostridium botulinum 14. Vibrio cholera 15. Mycobacterium bovis 16. Shigella species 17. Streptococcus pneumoniae 18. Clostridium perfringens 19. Salmonella typhi 20. Streptococcus pyogenes 3 II. Multiple Choice: Choose the ONE BEST answer. Mark the correct answer on Part 1 of the answer sheet. -

Compendium of Measures to Control Chlamydia Psittaci Infection Among
Compendium of Measures to Control Chlamydia psittaci Infection Among Humans (Psittacosis) and Pet Birds (Avian Chlamydiosis), 2017 Author(s): Gary Balsamo, DVM, MPH&TMCo-chair Angela M. Maxted, DVM, MS, PhD, Dipl ACVPM Joanne W. Midla, VMD, MPH, Dipl ACVPM Julia M. Murphy, DVM, MS, Dipl ACVPMCo-chair Ron Wohrle, DVM Thomas M. Edling, DVM, MSpVM, MPH (Pet Industry Joint Advisory Council) Pilar H. Fish, DVM (American Association of Zoo Veterinarians) Keven Flammer, DVM, Dipl ABVP (Avian) (Association of Avian Veterinarians) Denise Hyde, PharmD, RP Preeta K. Kutty, MD, MPH Miwako Kobayashi, MD, MPH Bettina Helm, DVM, MPH Brit Oiulfstad, DVM, MPH (Council of State and Territorial Epidemiologists) Branson W. Ritchie, DVM, MS, PhD, Dipl ABVP, Dipl ECZM (Avian) Mary Grace Stobierski, DVM, MPH, Dipl ACVPM (American Veterinary Medical Association Council on Public Health and Regulatory Veterinary Medicine) Karen Ehnert, and DVM, MPVM, Dipl ACVPM (American Veterinary Medical Association Council on Public Health and Regulatory Veterinary Medicine) Thomas N. Tully JrDVM, MS, Dipl ABVP (Avian), Dipl ECZM (Avian) (Association of Avian Veterinarians) Source: Journal of Avian Medicine and Surgery, 31(3):262-282. Published By: Association of Avian Veterinarians https://doi.org/10.1647/217-265 URL: http://www.bioone.org/doi/full/10.1647/217-265 BioOne (www.bioone.org) is a nonprofit, online aggregation of core research in the biological, ecological, and environmental sciences. BioOne provides a sustainable online platform for over 170 journals and books published by nonprofit societies, associations, museums, institutions, and presses. Your use of this PDF, the BioOne Web site, and all posted and associated content indicates your acceptance of BioOne’s Terms of Use, available at www.bioone.org/page/terms_of_use. -

2012 Case Definitions Infectious Disease
Arizona Department of Health Services Case Definitions for Reportable Communicable Morbidities 2012 TABLE OF CONTENTS Definition of Terms Used in Case Classification .......................................................................................................... 6 Definition of Bi-national Case ............................................................................................................................................. 7 ------------------------------------------------------------------------------------------------------- ............................................... 7 AMEBIASIS ............................................................................................................................................................................. 8 ANTHRAX (β) ......................................................................................................................................................................... 9 ASEPTIC MENINGITIS (viral) ......................................................................................................................................... 11 BASIDIOBOLOMYCOSIS ................................................................................................................................................. 12 BOTULISM, FOODBORNE (β) ....................................................................................................................................... 13 BOTULISM, INFANT (β) ................................................................................................................................................... -

World Health Orsanization Manila
REGIONAL OFFICE FOR THE WESTERN PACIFIC of the World Health Orsanization Manila REPORT ON THE SECOND REGIONAL SEMINAR ON VENEREAL DISEASE CONTROL MANILA. PHILIPPINES, 3 - 12 DECEMBER 1968 ., REGIONAL OFfiCE fOR THE WESTERN PACIFIC OF THE WORLD HEALTH ORGANIZATION MANILA ~PORl' ON rrHE SECOND REGIONAL SEMlNAR ON VENEREAL DISEASE CONTROL - Manila. Philippines 3 to 12 Decemher, 1968 WPRO 0144 SECOND RlOOIONAL SEMINAR ON VENEREAL DISEASE CONTBOL Sponsored by the WORLD HEALTH ORGANIZATION RIDIONAL OFFICE FOR THE WESTERN PACIFIC Manila, Philippines 3 to 12 December 1968 FINAL REPORT NOT FOR SALE PRINTED AND DISTRIBUTED by the REGIONAL OFFICE FOR THE WESTERN PACIFIC of the World Health Organization Manila, Philippines August 1969 CONTENTS PREFACE ~ 1. INTRODUCTION: THE CHANGING ENVIRONMENT •••••••••••••••••••••••• 1 2. NATURE AND ~ OF THE PBOBLEM .............................. 2 3. DIAGNOSIS OF VENEREAL DISEASES ....................•..••......•• 6 4.. TREA'D-mNT OF VENEREAL DlSEAS:e:f> ................................................................ .. 11 5.. VENEREAL DISEASE CONTROL .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. 17 6. BEHAVIOURAL PA'1"1'ERNS, HEALTH ElXJCATION AND ATTITUDES ........... 33 7 .. roruRE (J(]IJ!I.()OK .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. 35 8. SUMMARY AND RECOMMENDATIONS ............•..................•.... 35 9.. RE:P'ERmlCES .. .. . -

What Is Lyme Disease?
The threat of Lyme disease Dr Roger Evans Consultant Clinical Scientist National Lyme Borreliosis Testing Laboratory What is Lyme disease? • Also known as Lyme borreliosis • An infection caused by Borrelia burgdorferi sensu lato, a gram negative spirochaete bacterium • Transmitted through the bite of an infected tick (Ixodes ricinus in the UK) – a tick needs to be attached to the skin for around 24 hours to transmit Borrelia sp. to a person. • Recognised clinical presentations: early localized, early disseminated and late Lyme disease • There are large gaps in our clinical understanding of Lyme disease, particularly for ‘Post Treatment Lyme Disease Syndrome ‘ Picture credit: James Gathany Ixodes ricinus (tick) ecology/ life cycle Adapted from Manelli et al. 2011 Laboratory samples and cases of LB in Scotland 1996 to 2016 500 7000 450 6000 400 350 5000 300 4000 250 3000 200 Number of cases Number of samples 150 2000 100 1000 50 0 0 Cases Seroneg EM Year Samples GP study (2010-2012) • Three GP practices in NHS Highland region: Nairn, Culloden, Fort William • Number of laboratory cases compared to number of cases treated for Lyme disease without testing • About 2 x number of cases diagnosed by GP without lab testing compared to those that were tested • 2010-2011 • 1440 blood donors • Screened by EIA • EIA positive or equivocal samples confirmed by immunoblot (IB) • 60/1440 (4.2%) IB positive Munro et al (2015) Transfusion Medicine. Letter to the Editor doi: 10.1111/tme.12197 What does it cost? • Netherlands study (2010) – 5 health outcomes: -

Chlamydia Trachomatis Infection Is Driven by Nonprotective Immune Cells That Are Distinct from Protective Populations
Pathology after Chlamydia trachomatis infection is driven by nonprotective immune cells that are distinct from protective populations Rebeccah S. Lijeka,b,1, Jennifer D. Helblea, Andrew J. Olivea,c, Kyra W. Seigerb, and Michael N. Starnbacha,1 aDepartment of Microbiology and Immunobiology, Harvard Medical School, Boston, MA 02115; bDepartment of Biological Sciences, Mount Holyoke College, South Hadley, MA 01075; and cDepartment of Microbiology and Physiological Systems, University of Massachusetts Medical School, Worcester, MA 01605 Edited by Rafi Ahmed, Emory University, Atlanta, GA, and approved December 27, 2017 (received for review June 23, 2017) Infection with Chlamydia trachomatis drives severe mucosal immu- sequence identity, Chlamydia muridarum, the extent to which the nopathology; however, the immune responses that are required for molecular pathogenesis of C. muridarum represents that of Ct is mediating pathology vs. protection are not well understood. Here, unknown (6). Ct serovar L2 (Ct L2) is capable of infecting the we employed a mouse model to identify immune responses re- mouse upper genital tract when inoculated across the cervix into quired for C. trachomatis-induced upper genital tract pathology the uterus (7, 8) but it does not induce robust immunopathology. and to determine whether these responses are also required for This is consistent with the human disease phenotype caused by Ct L2, bacterial clearance. In mice as in humans, immunopathology was which disseminates to the lymph nodes causing lymphogranuloma characterized by extravasation of leukocytes into the upper genital venereum (LGV) and is not a major cause of mucosal immunopa- thology in the female upper genital tract (uterus and ovaries). tract that occluded luminal spaces in the uterus and ovaries. -

Practical Infection Control Guidelines
PRACTICAL INFECTION CONTROL GUIDELINES EDITION ONE CONTENTS INTRODUCTION 4 How to start 5 Four guiding principles 5 SECTION 1: HAND HYGIENE 6 Alcohol-based sanitisers 7 Hand washing 9 Factors that influence the effectiveness of hand hygiene 11 SECTION 2: PERSONAL PROTECTIVE EQUIPMENT 12 Laboratory coats/Scrubs 12 Non-sterile gowns 12 Gloves 13 Face protection 13 Respiratory protection 13 Footwear 14 Footbaths and foot mats 14 Table 1. Selection of appropriate protective equipment relative to risk 14 SECTION 3: ENVIRONMENTAL HYGIENE 15 Combining Cleaning and Disinfection 15 Cleaning 15 Disinfecting 16 Isolation Wards 16 Managing Patients in the isolation ward 17 Disinfectant selection 18 Table 2. Characteristics of selected disinfectants 19 Table 3. Commonly used disinfectants 20 Table 4. Antimicrobial spectrum of selected disinfectants 21 Miscellaneous items 21 SECTION 4: GENERAL PROCEDURES 22 Introduction 22 Cleaning of examination rooms 22 Cleaning of stethoscopes and smart devices 23 Cleaning of otoscopes 23 Cleaning of video-otoscopy units 23 Cleaning of diagnostic equipment (ultrasound machines, radiography machines) 23 Anaesthetic equipment disinfection 24 Cleaning of endoscopes 24 Endoscope disinfection with a liquid chemical agent involves five steps after leak testing 25 SECTION 4: GENERAL PROCEDURES CONTINUED 25 Surgery 25 Surgical Theatre 25 Personal Protective Equipment 25 Hand Hygiene 26 Preoperative-care 26 Skin Preparation 26 Post-operative care 26 Prophylactic antimicrobial use 26 Instrument sterilisation 27 Cold sterilisation using immersion in antiseptic solutions 27 Commonly performed high risk procedures 27 A. Otoscopic examination in a consult room 27 Instrument sterilisation 27 B. Ear flushing 28 Procedures area 28 Animal preparation 28 Personal Protective Equipment 28 Instrument sterilisation 29 C. -

Leptospirosis: a Waterborne Zoonotic Disease of Global Importance
August 2006 volume 22 number 08 Leptospirosis: A waterborne zoonotic disease of global importance INTRODUCTION syndrome has two phases: a septicemic and an immune phase (Levett, 2005). Leptospirosis is considered one of the most common zoonotic diseases It is in the immune phase that organ-specific damage and more severe illness globally. In the United States, outbreaks are increasingly being reported is seen. See text box for more information on the two phases. The typical among those participating in recreational water activities (Centers for Disease presenting signs of leptospirosis in humans are fever, headache, chills, con- Control and Prevention [CDC], 1996, 1998, and 2001) and sporadic cases are junctival suffusion, and myalgia (particularly in calf and lumbar areas) often underdiagnosed. With the onset of warm temperatures, increased (Heymann, 2004). Less common signs include a biphasic fever, meningitis, outdoor activities, and travel, Georgia may expect to see more leptospirosis photosensitivity, rash, and hepatic or renal failure. cases. DIAGNOSIS OF LEPTOSPIROSIS Leptospirosis is a zoonosis caused by infection with the bacterium Leptospira Detecting serum antibodies against leptospira interrogans. The disease occurs worldwide, but it is most common in temper- • Microscopic Agglutination Titers (MAT) ate regions in the late summer and early fall and in tropical regions during o Paired serum samples which show a four-fold rise in rainy seasons. It is not surprising that Hawaii has the highest incidence of titer confirm the diagnosis; a single high titer in a per- leptospirosis in the United States (Levett, 2005). The reservoir of pathogenic son clinically suspected to have leptospirosis is highly leptospires is the renal tubules of wild and domestic animals. -

Gonorrhea, Chlamydia, and Syphilis
2019 GONORRHEA, CHLAMYDIA, AND SYPHILIS AND CHLAMYDIA, GONORRHEA, Dedication TAG would like to thank the National Coalition of STD Directors for funding and input on the report. THE PIPELINE REPORT Pipeline for Gonorrhea, Chlamydia, and Syphilis By Jeremiah Johnson Introduction The current toolbox for addressing gonorrhea, chlamydia, and syphilis is inadequate. At a time where all three epidemics are dramatically expanding in locations all around the globe, including record-breaking rates of new infections in the United States, stakeholders must make do with old tools, inadequate systems for addressing sexual health, and a sparse research pipeline of new treatment, prevention, and diagnostic options. Lack of investment in sexual health research has left the field with inadequate prevention options, and limited access to infrastructure for testing and treatment have allowed sexually transmitted infections (STIs) to flourish. The consequences of this underinvestment are large: according to the World Health Organization (WHO), in 2012 there were an estimated 357 million new infections (roughly 1 million per day) of the four curable STIs: gonorrhea, chlamydia, syphilis, and trichomoniasis.1 In the United States, the three reportable STIs that are the focus of this report—gonorrhea, chlamydia, and syphilis—are growing at record paces. In 2017, a total of 30,644 cases of primary and secondary (P&S) syphilis—the most infectious stages of the disease—were reported in the United States. Since reaching a historic low in 2000 and 2001, the rate of P&S syphilis has increased almost every year, increasing 10.5% during 2016–2017. Also in 2017, 555,608 cases of gonorrhea were reported to the U.S. -
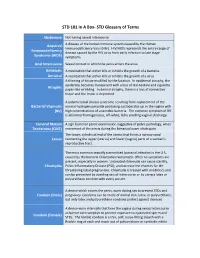
STD Glossary of Terms
STD 101 In A Box- STD Glossary of Terms Abstinence Not having sexual intercourse Acquired A disease of the human immune system caused by the Human Immunodeficiency Virus (HIV). HIV/AIDS represents the entire range of Immunodeficiency disease caused by the HIV virus from early infection to late stage Syndrome (AIDS) symptoms. Anal Intercourse Sexual contact in which the penis enters the anus. Antibiotic A medication that either kills or inhibits the growth of a bacteria. Antiviral A medication that either kills or inhibits the growth of a virus. A thinning of tissue modified by the location. In epidermal atrophy, the epidermis becomes transparent with a loss of skin texture and cigarette Atrophic paper-like wrinkling. In dermal atrophy, there is a loss of connective tissue and the lesion is depressed. A polymicrobial clinical syndrome resulting from replacement of the Bacterial Vaginosis normal hydrogen peroxide producing Lactobacillus sp. in the vagina with (BV) high concentrations of anaerobic bacteria. The common symptom of BV is abnormal homogeneous, off-white, fishy smelling vaginal discharge. Cervical Motion A sign found on pelvic examination suggestive of pelvic pathology; when Tenderness (CMT) movement of the cervix during the bimanual exam elicits pain. The lower, cylindrical end of the uterus that forms a narrow canal Cervix connecting the upper (uterus) and lower (vagina) parts of a woman's reproductive tract. The most common sexually transmitted bacterial infection in the U.S., caused by the bacteria Chlamydia trachomatis. Often no symptoms are present, especially in women. Untreated chlamydia can cause sterility, Chlamydia Pelvic Inflammatory Disease (PID), and increase the chances for life- threatening tubal pregnancies. -

Zoonotic Diseases Birds
Zoonotic Diseases Birds Zoonotic diseases Psittacosis (Ornithosis, Chlamydiosis): Psittacosis is caused by the bacteria Chlamydia psittaci. C. psittaci is common in wild birds and can occur in laboratory bird colonies. Infected birds are highly contagious to other birds and to humans. The organism is spread to humans by aerosolization of respiratory secretions or feces from the infected birds. Typical symptoms in the bird are diarrhea, ocular discharge, and nasal discharge. The infection in humans by C.psittaci, can cause fever, headache, myalgia chills, and upper and lower respiratory disease. Serious complications can occur and include pneumonia, hepatitis, myocarditis, thrombophlebitis and encephalitis. It is responsive to antibiotic therapy but relapses can occur in untreated infections. Prevention: Only disease-free flocks should be allowed into the research facility. Wild-caught birds or birds of unknown status should be treated prophylactically for 45 days with chlortetracycline. Animal Biosafety Level 2 practices are recommended for personnel working with naturally infected birds or experimentally infected birds. Wearing NIOSH certified dust masks should be considered in rooms housing birds of unknown health status. Newcastle Disease: Newcastle disease is caused by a paramyxovirus and can be seen in birds both wild and domestic. Transmission is mainly by aerosol but contaminated food, water and equipment can also transmit the infection within bird colonies. Pathogenic strains produce anorexia and respiratory disease in adult birds.Young birds often show neurologic signs. In humans the disease is characterized by conjunctivitis, fever, and respiratory symptoms. Prevention: The disease can be prevented by immunizing susceptible birds and obtaining birds from flocks free of infection.