2-Tesis LG-Correcciones Censor
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

DIAGMOL Town, ZIPCODE :…………………………………………………… Director : Prof
Mr. Mrs. (IN UPPER CASE, please) Name:……………………………………………………. Maiden Name:……………………………………. DMGL / Service de Médecine Génétique Centre d’accueil des prélèvements (CAP) First name :………………………………………………… Bâtiment des Laboratoires (BATLab), local 8D-0-850.1 Date of birth : ............ / ........... / …………… 4 rue Gabrielle-Perret-Gentil, 1211 Genève 14 Legal representative (for minors) : father mother Molecular and Genomic Diagnostics Laboratory Name/first name :…………………………………………… Street/N°:……………………………………………………… http://www.hug-ge.ch/feuilles-de-demande DIAGMOL Town, ZIPCODE :…………………………………………………… Director : Prof. Stylianos ANTONARAKIS Hospitalisation Unit: …………… Physician :…………………… Lab managers: N° EdS : …………………………………………………………… Dr J.-L. BLOUIN, Dr Th. NOUSPIKEL, Dr. M. GUIPPONI Invoice address: Patient Prescriptor Insurance [email protected], [email protected], [email protected] Type of case : Disease AI Accident Pregnancy Lab direct or results: Phone/FAX: +41 (0) 22 37 21 826 / 21 860 N° AVS (Mandatory for AI) : ………………………...................... Sample Entrance Center (CAP) : Phone +41 (0) 22 37 21 800 Insurance : ………………… Insured N° : ……………………… PHYSICIAN PHYSICIAN (NAME/First name - Street/N°- Town, ZIPCODE - Phone/FAX. IN UPPER CASES, PLEASE) COPY TO OTHER PHYSICIAN (NAME/First name - Street/N°- Town, ZIPCODE - Phone/FAX. IN UPPER CASES, PLEASE) « The laboratory is granted permission by the Physician/Patient to transmit copies of the report to other physicians» Opposition of the patient to the registration of this request results in the electronic patient record (DPI) of the HUG If the patient belongs to a family already known to the laboratory, please indicate index case NAME: CLINICAL INFORMATIONS given by the physician: Ethnic origins Father Mother Currently pregnant Date of last menses Number of weeks of amenorrhea SAMPLE(S) Most of our tests work from 4 ml of blood in EDTA (children <2 ans : 1 On every and single NAME First name ml : ok) or from purified DNA (some exceptions apply for some tests) tube. -

University of California, San Diego
UNIVERSITY OF CALIFORNIA, SAN DIEGO The post-terminal differentiation fate of RNAs revealed by next-generation sequencing A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Biomedical Sciences by Gloria Kuo Lefkowitz Committee in Charge: Professor Benjamin D. Yu, Chair Professor Richard Gallo Professor Bruce A. Hamilton Professor Miles F. Wilkinson Professor Eugene Yeo 2012 Copyright Gloria Kuo Lefkowitz, 2012 All rights reserved. The Dissertation of Gloria Kuo Lefkowitz is approved, and it is acceptable in quality and form for publication on microfilm and electronically: __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ Chair University of California, San Diego 2012 iii DEDICATION Ma and Ba, for your early indulgence and support. Matt and James, for choosing more practical callings. Roy, my love, for patiently sharing the ups and downs of this journey. iv EPIGRAPH It is foolish to tear one's hair in grief, as though sorrow would be made less by baldness. ~Cicero v TABLE OF CONTENTS Signature Page .............................................................................................................. iii Dedication .................................................................................................................... -

Differential Epigenomic and Transcriptomic Responses in Subcutaneous Adipose Tissue Between Low and High Responders to Caloric Restriction1–3
Differential epigenomic and transcriptomic responses in subcutaneous adipose tissue between low and high responders to caloric restriction1–3 Luigi Bouchard, Re´mi Rabasa-Lhoret, May Faraj, Marie-E`ve Lavoie, Jonathan Mill, Louis Pe´russe, and Marie-Claude Vohl ABSTRACT weight loss responses to caloric restriction show considerable Background: Caloric restriction is recommended for the treatment interindividual variability (7). Studies of genetically identical of obesity, but it is generally characterized by large interindividual monozygotic twins have been particularly useful in disentangling variability in responses. The factors affecting the magnitude of the role of environmental and heritable factors in determining the weight loss remain poorly understood. Epigenetic factors (ie, heri- degree of weight loss. It has been shown that within-pair changes table but reversible changes to genomic function that regulate gene in body fat variability after a caloric deficit is significantly lower expression independently of DNA sequence) may explain some of than between-pair variability, which suggests that genetic factors the interindividual variability seen in weight-loss responses. have an important influence on an individual’s response to caloric Objective: The objective was to determine whether epigenetics and deficit (8, 9). However, the concordance between twin pairs was gene expression changes may play a role in weight-loss responsiveness. not complete, which suggests that environmental factors or other Design: Overweight/obese postmenopausal women were recruited DNA sequence–independent mechanisms may be involved. for a standard 6-mo caloric restriction intervention. Abdominal sub- It has been suggested that monozygotic twin discordance for cutaneous adipose tissue biopsy samples were collected before (n = complex traits such as body weight could be accounted for by 14) and after (n = 14) intervention, and the epigenomic and tran- epigenetic factors (10, 11). -

Identification of Novel Sleep Related Genes from Large Scale Phenotyping Experiments in Mice
University of Kentucky UKnowledge Theses and Dissertations--Biology Biology 2017 IDENTIFICATION OF NOVEL SLEEP RELATED GENES FROM LARGE SCALE PHENOTYPING EXPERIMENTS IN MICE Shreyas Joshi University of Kentucky, [email protected] Digital Object Identifier: https://doi.org/10.13023/ETD.2017.159 Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Recommended Citation Joshi, Shreyas, "IDENTIFICATION OF NOVEL SLEEP RELATED GENES FROM LARGE SCALE PHENOTYPING EXPERIMENTS IN MICE" (2017). Theses and Dissertations--Biology. 42. https://uknowledge.uky.edu/biology_etds/42 This Doctoral Dissertation is brought to you for free and open access by the Biology at UKnowledge. It has been accepted for inclusion in Theses and Dissertations--Biology by an authorized administrator of UKnowledge. For more information, please contact [email protected]. STUDENT AGREEMENT: I represent that my thesis or dissertation and abstract are my original work. Proper attribution has been given to all outside sources. I understand that I am solely responsible for obtaining any needed copyright permissions. I have obtained needed written permission statement(s) from the owner(s) of each third-party copyrighted matter to be included in my work, allowing electronic distribution (if such use is not permitted by the fair use doctrine) which will be submitted to UKnowledge as Additional File. I hereby grant to The University of Kentucky and its agents the irrevocable, non-exclusive, and royalty-free license to archive and make accessible my work in whole or in part in all forms of media, now or hereafter known. -

Literature Mining Sustains and Enhances Knowledge Discovery from Omic Studies
LITERATURE MINING SUSTAINS AND ENHANCES KNOWLEDGE DISCOVERY FROM OMIC STUDIES by Rick Matthew Jordan B.S. Biology, University of Pittsburgh, 1996 M.S. Molecular Biology/Biotechnology, East Carolina University, 2001 M.S. Biomedical Informatics, University of Pittsburgh, 2005 Submitted to the Graduate Faculty of School of Medicine in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Pittsburgh 2016 UNIVERSITY OF PITTSBURGH SCHOOL OF MEDICINE This dissertation was presented by Rick Matthew Jordan It was defended on December 2, 2015 and approved by Shyam Visweswaran, M.D., Ph.D., Associate Professor Rebecca Jacobson, M.D., M.S., Professor Songjian Lu, Ph.D., Assistant Professor Dissertation Advisor: Vanathi Gopalakrishnan, Ph.D., Associate Professor ii Copyright © by Rick Matthew Jordan 2016 iii LITERATURE MINING SUSTAINS AND ENHANCES KNOWLEDGE DISCOVERY FROM OMIC STUDIES Rick Matthew Jordan, M.S. University of Pittsburgh, 2016 Genomic, proteomic and other experimentally generated data from studies of biological systems aiming to discover disease biomarkers are currently analyzed without sufficient supporting evidence from the literature due to complexities associated with automated processing. Extracting prior knowledge about markers associated with biological sample types and disease states from the literature is tedious, and little research has been performed to understand how to use this knowledge to inform the generation of classification models from ‘omic’ data. Using pathway analysis methods to better understand the underlying biology of complex diseases such as breast and lung cancers is state-of-the-art. However, the problem of how to combine literature- mining evidence with pathway analysis evidence is an open problem in biomedical informatics research. -

UNSCEAR 2001 Report to the General Assembly, with Scientific Annex
HEREDITARY EFFECTS OF RADIATION United Nations Scientific Committee on the Effects of Atomic Radiation UNSCEAR 2001 Report to the General Assembly, with Scientific Annex UNITED NATIONS HEREDITARY EFFECTS OF RADIATION United Nations Scientific Committee on the Effects of Atomic Radiation UNSCEAR 2001 Report to the General Assembly, with Scientific Annex UNITED NATIONS New York, 2001 NOTE The report of the Committee without its scientific annex appears as Official Records of the General Assembly. Fifty-sixth Session, Supplement No. 46 (N56146). The designation employed and the presentation of material in this publication do not imply the expression of any opinion whatsoever on the pad of the Secretariat of the United Nationsconcerning the legal status of any country, temtory, city orarea, or of its authorities, or concerning the delimitation of its frontiers or boundaries. The country names used in this document are, In most cases, those that were in use at the time the data were collected or the text prepared. In othercases, however, the names have been updated, where this was posslble and appropriate, to reflect political changes. UNITED NATIONS PUBLICATION Sales No. E.O1 .IX.2 ISBN 92-1-1 42244-2 Report of the United Nations Scientific Committee on the Effects of Atomic Radiation to the General Assembly 1. During the past few years, the United Nations 4. The Committee wishes to acknowledge the assistance Scientific Committee on the Effects of Atomic Radiation1 of the consultant, K. Sankaranarayanan, in the preparation has undertaken broad reviews of the sources and effects of of the scientific annex and the advice of the international ionizing radiation. -

Uncommon Forms of Diabetes
Clinical Medicine 2021 Vol 21, No 4: e337–41 CME: DIABETES Uncommon forms of diabetes Authors: Yun-Ni LeeA and Mohammed SB HudaB Diabetes mellitus is a common condition which all clinicians and insulin independence. It is estimated to account for 1%–2% will encounter in their clinical practice. The most common of patients diagnosed with diabetes and, in the UK, the prevalence form is type 2 diabetes followed by type 1 diabetes. However, of MODY is estimated to be at 108 cases per million.3 However, there are many other atypical forms of diabetes which are it may be a significant underestimate and these figures are not important for a clinician to consider as it can impact on the accurate until large population screening studies are performed. ABSTRACT diagnosis and their management. The most common mutations are hepatocyte nuclear factor-1- This article focuses on maturity onset diabetes of the young alpha (HNF1α; 52%), glucokinase (GCK; 32%) and HNF4α (10%), (MODY), latent autoimmune diabetes in adults (LADA), see Table 2.3 ketosis-prone diabetes and other secondary forms of diabetes such as pancreatic cancer and haemochromatosis. We briefly Hepatocyte nuclear factor-1-alpha gene describe the key clinical features of these forms of diabetes and their investigations and treatment. Formerly called MODY3, mutations on the HNF1α gene on chromosome 3 are associated with a progressive defect of insulin secretion.4 Mutations here also result in low renal threshold for 5 Introduction glucose and thus mutation carriers have detectable glycosuria. In the UK, around 90% of people with diabetes have type 2 diabetes (T2D), around 8% have type 1 diabetes (T1D) and around 2% have other forms of diabetes.1 Key points Typically, we see T1D present in a young, lean patient with Suspect other uncommon forms of diabetes if the clinical marked symptoms of polyuria, polydipsia, weight loss and diabetic picture does not fit type 1 or type 2 diabetes. -

Mat Kadi Tora Tutti O Al Ut Hit Hitta Atuh
MAT KADI TORA TUTTI USO AL20180235194A1 UT HIT HITTA ATUH ( 19) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2018 /0235194 A1 Fahrenkrug et al. (43 ) Pub . Date : Aug . 23, 2018 ( 54 ) MULTIPLEX GENE EDITING Publication Classification (51 ) Int. Ci. ( 71 ) Applicant: Recombinetics , Inc ., Saint Paul, MN A01K 67/ 027 (2006 . 01 ) (US ) C12N 15 / 90 ( 2006 .01 ) (72 ) Inventors : Scott C . Fahrenkrug, Minneapolis , (52 ) U . S . CI. MN (US ) ; Daniel F . Carlson , CPC .. .. A01K 67 / 0276 (2013 . 01 ) ; C12N 15 / 907 Woodbury , MN (US ) ( 2013 .01 ) ; A01K 67 /0275 ( 2013 .01 ) ; A01K 2267/ 02 (2013 .01 ) ; AOIK 2217 / 15 (2013 .01 ) ; AOIK 2227 / 108 ( 2013 .01 ) ; AOIK 2217 /07 (21 ) Appl. No. : 15 /923 , 951 ( 2013 .01 ) ; A01K 2227/ 101 (2013 .01 ) ; AOIK ( 22 ) Filed : Mar. 16 , 2018 2217 /075 ( 2013 .01 ) (57 ) ABSTRACT Related U . S . Application Data Materials and methods for making multiplex gene edits in (62 ) Division of application No . 14 /698 ,561 , filed on Apr. cells and are presented . Further methods include animals 28 , 2015, now abandoned . and methods of making the same . Methods of making ( 60 ) Provisional application No . 61/ 985, 327, filed on Apr. chimeric animals are presented , as well as chimeric animals . 28 , 2014 . Specification includes a Sequence Listing . Patent Application Publication Aug . 23 , 2018 Sheet 1 of 13 US 2018 / 0235194 A1 GENERATION OF HOMOZYGOUS CATTLE EDITED AT ONE ALLELE USING SINGLE EDITS Edit allele , Raise FO to Mate FO enough times Raise F1s to Mate F1 siblings Clone cell , sexual maturity , to produce enough F1 sexualmaturity , to make Implant, Gestate 2 years generation carrying 2 years homozygous KO , 9 months , birth edited allele to mate 9 months of FO with each other Generation Primary Fibroblasts ? ? ? Time, years FIG . -

University of California, San Diego
UC San Diego UC San Diego Electronic Theses and Dissertations Title The post-terminal differentiation fate of RNAs revealed by next-generation sequencing Permalink https://escholarship.org/uc/item/7324r1rj Author Lefkowitz, Gloria Kuo Publication Date 2012 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA, SAN DIEGO The post-terminal differentiation fate of RNAs revealed by next-generation sequencing A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Biomedical Sciences by Gloria Kuo Lefkowitz Committee in Charge: Professor Benjamin D. Yu, Chair Professor Richard Gallo Professor Bruce A. Hamilton Professor Miles F. Wilkinson Professor Eugene Yeo 2012 Copyright Gloria Kuo Lefkowitz, 2012 All rights reserved. The Dissertation of Gloria Kuo Lefkowitz is approved, and it is acceptable in quality and form for publication on microfilm and electronically: __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ Chair University of California, San Diego 2012 iii DEDICATION Ma and Ba, for your early indulgence and support. Matt and James, for choosing more practical callings. Roy, my love, for patiently sharing the ups and downs -
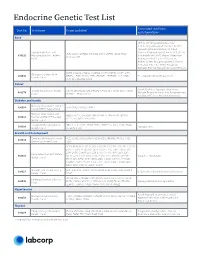
Endocrine Genetic Test List
Endocrine Genetic Test List Associated conditions Test No. Test Name Genes Included and phenotypes Bone HPP, X-Linked Hypophosphatemia, X-Linked Hypophosphatemic Rickets, XLH, Hypophosphatemic Rickets, X-Linked Hypophosphatasia and Dominant Hypophosphatemic Rickets (XLHR), ALPL, CLCN5, CYP2R1, CYP27B1, DMP1, ENPP1, FGF23, PHEX, 630292 Hypophosphatemic Rickets X-Linked Rickets (XLR), Vitamin D-Resistant SLC34A3, VDR Panel Rickets, X-Linked Vitamin D-Resistant Rickets (VDRR), Hypophosphatemic Vitamin D-Resistant Rickets (HPDR), Phosphate Diabetes, Familial Hypophosphatemic Rickets BMP1, COL1A1, COL1A2, CREB3L1, CRTAP, FKBP10, IFITM5, LRP5, Osteogenesis Imperfecta 630543 MBTPS2, P3H1, PLOD2, PPIB, SERPINF1, SERPINH1, SP7, SPARC, OI, Juvenile Primary Osteoporosis Genetic Panel TENT5A, TMEM38B, WNT1 Cancer Familial Isolated Hyperparathyroidism, VistaSeq® Endocrine Cancer CDC73, MAX, MEN1, NF1, PRKAR1A, PTEN, RET, SDHB, SDHC, SDHD, 481374 Multiple Endocrine Neoplasia, Paraganglioma, Panel* TMEM127, TP53 and VHL Parathyroid Cancer, Pheochromocytoma Diabetes and Insulin Maturity-Onset Diabetes of the 630568 GCK, HNF1A, HNF1B, HNF4A Young (MODY) 4-gene Panel Maturity-Onset Diabetes of ABCC8, APPL1, BLK, GCK, HNF1A, HNF1B, HNF4A, INS, KCNJ11, 630513 the Young (MODY) Expanded KLF11, NEUROD1, PAX4, PDX1 Genetic Panel Congenital Hyperinsulinism ABCC8, GCK, GLUD1, HADH, HNF1A, HNF4A, KCNJ11, PGM1, PMM2, 630500 Hypoglycemia Genetic Panel SLC16A1, UCP2 Growth and Development Combined Pituitary Hormone GLI1, HESX1, LHX3, LHX4, OTX2, POU1F1, PROKR2, PROP1, -

DNASE1L2 (A-14): Sc-68312
SAN TA C RUZ BI OTEC HNOL OG Y, INC . DNASE1L2 (A-14): sc-68312 BACKGROUND APPLICATIONS DNASE1L2 (deoxyribonuclease I-like 2), also known as DHP1 or DNAS1L2, is a DNASE1L2 (A-14) is recommended for detection of DNASE1L2 of human 299 amino acid secreted protein that is expressed in brain tissue and shares origin by Western Blotting (starting dilution 1:200, dilution range 1:100- sequence similarity with DNase I, suggesting a possibly role in DNA hydroly - 1:1000), immunoprecipitation [1-2 µg per 100-500 µg of total protein (1 ml sis. The gene encoding DNASE1L2 maps to human chromosome 16, which of cell lysate)], immunofluorescence (starting dilution 1:50, dilution range encodes over 900 genes and comprises nearly 3% of the human genome. The 1:50-1:500) and solid phase ELISA (starting dilution 1:30, dilution range GAN gene is located on chromosome 16 and, with mutation, may lead to giant 1:30- 1:3000). axonal neuropathy, a nervous system disorder characterized by increasing Suitable for use as control antibody for DNASE1L2 siRNA (h): sc-77320, malfunction with growth. The rare disorder Rubinstein-Taybi syndrome is also DNASE1L2 shRNA Plasmid (h): sc-77320-SH and DNASE1L2 shRNA (h) associated with chromosome 16, as is Crohn’s disease, which is a gastroin - Lentiviral Particles: sc-77320-V. testinal inflammatory condition. Molecular Weight (predicted) of DNASE1L2: 33 kDa. REFERENCES Molecular Weight (observed) of DNASE1L2: 37 kDa. 1. Rodriguez, A.M., Rodin, D., Nomura, H., Morton, C.C., Weremowicz, S. and Positive Controls: IMR-32 nuclear extract: sc-2148. -

An Interesting Case of Young Onset Diabetes Mellitus
International Journal of Research in Medical Sciences Gulati S et al. Int J Res Med Sci. 2017 Sep;5(9):4178-4180 www.msjonline.org pISSN 2320-6071 | eISSN 2320-6012 DOI: http://dx.doi.org/10.18203/2320-6012.ijrms20174007 Case Report An interesting case of young onset diabetes mellitus Shipra Gulati*, Tanvi Batra, Akshay A. Dhamne, Vijayashree S. Gokhale Department of Medicine, Dr. DY Patil Medical College and Hospital, Pimpri, Pune, Maharashtra, India Received: 23 June 2017 Accepted: 22 July 2017 *Correspondence: Dr. Shipra Gulati, E-mail: [email protected] Copyright: © the author(s), publisher and licensee Medip Academy. This is an open-access article distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited. ABSTRACT A 24 years old female, was admitted with symptoms of urinary tract infection. She was married and had bad obstetric history. She was known diabetic for 16 years of age and was on regular treatment with injection human insulin mixtard since the time of diagnosis, but had no episode of diabetic ketosis/ ketoacidosis. She had a positive family history of diabetes. She was further evaluated and was found to have normal C peptide levels and islet cell antibodies were found to be negative. Hence, the possibility of MODY (monogenic diabetes) was considered. Her genetic testing could not be done due to financial constraints. But a trial of sulfonylureas was given along with reduction in the dose of insulin to which she responded well and is presently well controlled.