UNSCEAR 2001 Report to the General Assembly, with Scientific Annex
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

DIAGMOL Town, ZIPCODE :…………………………………………………… Director : Prof
Mr. Mrs. (IN UPPER CASE, please) Name:……………………………………………………. Maiden Name:……………………………………. DMGL / Service de Médecine Génétique Centre d’accueil des prélèvements (CAP) First name :………………………………………………… Bâtiment des Laboratoires (BATLab), local 8D-0-850.1 Date of birth : ............ / ........... / …………… 4 rue Gabrielle-Perret-Gentil, 1211 Genève 14 Legal representative (for minors) : father mother Molecular and Genomic Diagnostics Laboratory Name/first name :…………………………………………… Street/N°:……………………………………………………… http://www.hug-ge.ch/feuilles-de-demande DIAGMOL Town, ZIPCODE :…………………………………………………… Director : Prof. Stylianos ANTONARAKIS Hospitalisation Unit: …………… Physician :…………………… Lab managers: N° EdS : …………………………………………………………… Dr J.-L. BLOUIN, Dr Th. NOUSPIKEL, Dr. M. GUIPPONI Invoice address: Patient Prescriptor Insurance [email protected], [email protected], [email protected] Type of case : Disease AI Accident Pregnancy Lab direct or results: Phone/FAX: +41 (0) 22 37 21 826 / 21 860 N° AVS (Mandatory for AI) : ………………………...................... Sample Entrance Center (CAP) : Phone +41 (0) 22 37 21 800 Insurance : ………………… Insured N° : ……………………… PHYSICIAN PHYSICIAN (NAME/First name - Street/N°- Town, ZIPCODE - Phone/FAX. IN UPPER CASES, PLEASE) COPY TO OTHER PHYSICIAN (NAME/First name - Street/N°- Town, ZIPCODE - Phone/FAX. IN UPPER CASES, PLEASE) « The laboratory is granted permission by the Physician/Patient to transmit copies of the report to other physicians» Opposition of the patient to the registration of this request results in the electronic patient record (DPI) of the HUG If the patient belongs to a family already known to the laboratory, please indicate index case NAME: CLINICAL INFORMATIONS given by the physician: Ethnic origins Father Mother Currently pregnant Date of last menses Number of weeks of amenorrhea SAMPLE(S) Most of our tests work from 4 ml of blood in EDTA (children <2 ans : 1 On every and single NAME First name ml : ok) or from purified DNA (some exceptions apply for some tests) tube. -

Abstracts from the 9Th Biennial Scientific Meeting of The
International Journal of Pediatric Endocrinology 2017, 2017(Suppl 1):15 DOI 10.1186/s13633-017-0054-x MEETING ABSTRACTS Open Access Abstracts from the 9th Biennial Scientific Meeting of the Asia Pacific Paediatric Endocrine Society (APPES) and the 50th Annual Meeting of the Japanese Society for Pediatric Endocrinology (JSPE) Tokyo, Japan. 17-20 November 2016 Published: 28 Dec 2017 PS1 Heritable forms of primary bone fragility in children typically lead to Fat fate and disease - from science to global policy a clinical diagnosis of either osteogenesis imperfecta (OI) or juvenile Peter Gluckman osteoporosis (JO). OI is usually caused by dominant mutations affect- Office of Chief Science Advsor to the Prime Minister ing one of the two genes that code for two collagen type I, but a re- International Journal of Pediatric Endocrinology 2017, 2017(Suppl 1):PS1 cessive form of OI is present in 5-10% of individuals with a clinical diagnosis of OI. Most of the involved genes code for proteins that Attempts to deal with the obesity epidemic based solely on adult be- play a role in the processing of collagen type I protein (BMP1, havioural change have been rather disappointing. Indeed the evidence CREB3L1, CRTAP, LEPRE1, P4HB, PPIB, FKBP10, PLOD2, SERPINF1, that biological, developmental and contextual factors are operating SERPINH1, SEC24D, SPARC, from the earliest stages in development and indeed across generations TMEM38B), or interfere with osteoblast function (SP7, WNT1). Specific is compelling. The marked individual differences in the sensitivity to the phenotypes are caused by mutations in SERPINF1 (recessive OI type obesogenic environment need to be understood at both the individual VI), P4HB (Cole-Carpenter syndrome) and SEC24D (‘Cole-Carpenter and population level. -

Abstracts from the 50Th European Society of Human Genetics Conference: Electronic Posters
European Journal of Human Genetics (2019) 26:820–1023 https://doi.org/10.1038/s41431-018-0248-6 ABSTRACT Abstracts from the 50th European Society of Human Genetics Conference: Electronic Posters Copenhagen, Denmark, May 27–30, 2017 Published online: 1 October 2018 © European Society of Human Genetics 2018 The ESHG 2017 marks the 50th Anniversary of the first ESHG Conference which took place in Copenhagen in 1967. Additional information about the event may be found on the conference website: https://2017.eshg.org/ Sponsorship: Publication of this supplement is sponsored by the European Society of Human Genetics. All authors were asked to address any potential bias in their abstract and to declare any competing financial interests. These disclosures are listed at the end of each abstract. Contributions of up to EUR 10 000 (ten thousand euros, or equivalent value in kind) per year per company are considered "modest". Contributions above EUR 10 000 per year are considered "significant". 1234567890();,: 1234567890();,: E-P01 Reproductive Genetics/Prenatal and fetal echocardiography. The molecular karyotyping Genetics revealed a gain in 8p11.22-p23.1 region with a size of 27.2 Mb containing 122 OMIM gene and a loss in 8p23.1- E-P01.02 p23.3 region with a size of 6.8 Mb containing 15 OMIM Prenatal diagnosis in a case of 8p inverted gene. The findings were correlated with 8p inverted dupli- duplication deletion syndrome cation deletion syndrome. Conclusion: Our study empha- sizes the importance of using additional molecular O¨. Kırbıyık, K. M. Erdog˘an, O¨.O¨zer Kaya, B. O¨zyılmaz, cytogenetic methods in clinical follow-up of complex Y. -

Uncommon Forms of Diabetes
Clinical Medicine 2021 Vol 21, No 4: e337–41 CME: DIABETES Uncommon forms of diabetes Authors: Yun-Ni LeeA and Mohammed SB HudaB Diabetes mellitus is a common condition which all clinicians and insulin independence. It is estimated to account for 1%–2% will encounter in their clinical practice. The most common of patients diagnosed with diabetes and, in the UK, the prevalence form is type 2 diabetes followed by type 1 diabetes. However, of MODY is estimated to be at 108 cases per million.3 However, there are many other atypical forms of diabetes which are it may be a significant underestimate and these figures are not important for a clinician to consider as it can impact on the accurate until large population screening studies are performed. ABSTRACT diagnosis and their management. The most common mutations are hepatocyte nuclear factor-1- This article focuses on maturity onset diabetes of the young alpha (HNF1α; 52%), glucokinase (GCK; 32%) and HNF4α (10%), (MODY), latent autoimmune diabetes in adults (LADA), see Table 2.3 ketosis-prone diabetes and other secondary forms of diabetes such as pancreatic cancer and haemochromatosis. We briefly Hepatocyte nuclear factor-1-alpha gene describe the key clinical features of these forms of diabetes and their investigations and treatment. Formerly called MODY3, mutations on the HNF1α gene on chromosome 3 are associated with a progressive defect of insulin secretion.4 Mutations here also result in low renal threshold for 5 Introduction glucose and thus mutation carriers have detectable glycosuria. In the UK, around 90% of people with diabetes have type 2 diabetes (T2D), around 8% have type 1 diabetes (T1D) and around 2% have other forms of diabetes.1 Key points Typically, we see T1D present in a young, lean patient with Suspect other uncommon forms of diabetes if the clinical marked symptoms of polyuria, polydipsia, weight loss and diabetic picture does not fit type 1 or type 2 diabetes. -

Genetic, Metabolic and Clinical Characteristics of Maturity Onset Diabetes of the Young
European Journal of Endocrinology (1998) 138 233±239 ISSN 0804-4643 REVIEW Genetic, metabolic and clinical characteristics of maturity onset diabetes of the young Gilberto Velho and Philippe Froguel1 INSERM U-342, HoÃpital Saint Vincent de Paul, Paris, France and 1CNRS EP10, Institut Pasteur de Lille et CHU, Lille, France (Correspondence should be addressed to G Velho, INSERM U-342, HoÃpital Saint Vincent de Paul, 82 Avenue Denfert Rochereau, 75014 Paris, France) Abstract Maturity onset diabetes of the young (MODY) is a genetically and clinically heterogeneous subtype of non-insulin-dependent diabetes mellitus (NIDDM) characterised by early onset, autosomal dominant inheritance and a primary defect in insulin secretion. To date, three MODY genes have been identi®ed on chromosomes 20q (MODY1/hepatic nuclear factor (HNF)-4a), 7p (MODY2/glucokinase) and 12q (MODY3/HNF-1a). Mutations in MODY2/glucokinase result in mild chronic hyperglycaemia as a result of reduced pancreatic beta-cell responsiveness to glucose, and decreased net accumulation of hepatic glycogen and increased hepatic gluconeogenesis after meals. In contrast, MODY1 and MODY3 are characterised by severe insulin secretory defects, and by major hyperglycaemia associated with microvascular complications. The role of the three known MODY genes in susceptibility to the more common late-onset NIDDM remain uncertain. Genetic studies seem to exclude a role as major susceptibility genes, but leave unresolved whether they may have a minor role in a polygenic context or an important role in particular populations. European Journal of Endocrinology 138 233±239 MODY is a monogenic form of NIDDM MODY3/HNF-1a (11, 12) respectively, can cause this form of diabetes. -

Mat Kadi Tora Tutti O Al Ut Hit Hitta Atuh
MAT KADI TORA TUTTI USO AL20180235194A1 UT HIT HITTA ATUH ( 19) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2018 /0235194 A1 Fahrenkrug et al. (43 ) Pub . Date : Aug . 23, 2018 ( 54 ) MULTIPLEX GENE EDITING Publication Classification (51 ) Int. Ci. ( 71 ) Applicant: Recombinetics , Inc ., Saint Paul, MN A01K 67/ 027 (2006 . 01 ) (US ) C12N 15 / 90 ( 2006 .01 ) (72 ) Inventors : Scott C . Fahrenkrug, Minneapolis , (52 ) U . S . CI. MN (US ) ; Daniel F . Carlson , CPC .. .. A01K 67 / 0276 (2013 . 01 ) ; C12N 15 / 907 Woodbury , MN (US ) ( 2013 .01 ) ; A01K 67 /0275 ( 2013 .01 ) ; A01K 2267/ 02 (2013 .01 ) ; AOIK 2217 / 15 (2013 .01 ) ; AOIK 2227 / 108 ( 2013 .01 ) ; AOIK 2217 /07 (21 ) Appl. No. : 15 /923 , 951 ( 2013 .01 ) ; A01K 2227/ 101 (2013 .01 ) ; AOIK ( 22 ) Filed : Mar. 16 , 2018 2217 /075 ( 2013 .01 ) (57 ) ABSTRACT Related U . S . Application Data Materials and methods for making multiplex gene edits in (62 ) Division of application No . 14 /698 ,561 , filed on Apr. cells and are presented . Further methods include animals 28 , 2015, now abandoned . and methods of making the same . Methods of making ( 60 ) Provisional application No . 61/ 985, 327, filed on Apr. chimeric animals are presented , as well as chimeric animals . 28 , 2014 . Specification includes a Sequence Listing . Patent Application Publication Aug . 23 , 2018 Sheet 1 of 13 US 2018 / 0235194 A1 GENERATION OF HOMOZYGOUS CATTLE EDITED AT ONE ALLELE USING SINGLE EDITS Edit allele , Raise FO to Mate FO enough times Raise F1s to Mate F1 siblings Clone cell , sexual maturity , to produce enough F1 sexualmaturity , to make Implant, Gestate 2 years generation carrying 2 years homozygous KO , 9 months , birth edited allele to mate 9 months of FO with each other Generation Primary Fibroblasts ? ? ? Time, years FIG . -
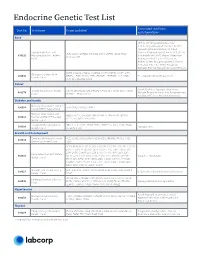
Endocrine Genetic Test List
Endocrine Genetic Test List Associated conditions Test No. Test Name Genes Included and phenotypes Bone HPP, X-Linked Hypophosphatemia, X-Linked Hypophosphatemic Rickets, XLH, Hypophosphatemic Rickets, X-Linked Hypophosphatasia and Dominant Hypophosphatemic Rickets (XLHR), ALPL, CLCN5, CYP2R1, CYP27B1, DMP1, ENPP1, FGF23, PHEX, 630292 Hypophosphatemic Rickets X-Linked Rickets (XLR), Vitamin D-Resistant SLC34A3, VDR Panel Rickets, X-Linked Vitamin D-Resistant Rickets (VDRR), Hypophosphatemic Vitamin D-Resistant Rickets (HPDR), Phosphate Diabetes, Familial Hypophosphatemic Rickets BMP1, COL1A1, COL1A2, CREB3L1, CRTAP, FKBP10, IFITM5, LRP5, Osteogenesis Imperfecta 630543 MBTPS2, P3H1, PLOD2, PPIB, SERPINF1, SERPINH1, SP7, SPARC, OI, Juvenile Primary Osteoporosis Genetic Panel TENT5A, TMEM38B, WNT1 Cancer Familial Isolated Hyperparathyroidism, VistaSeq® Endocrine Cancer CDC73, MAX, MEN1, NF1, PRKAR1A, PTEN, RET, SDHB, SDHC, SDHD, 481374 Multiple Endocrine Neoplasia, Paraganglioma, Panel* TMEM127, TP53 and VHL Parathyroid Cancer, Pheochromocytoma Diabetes and Insulin Maturity-Onset Diabetes of the 630568 GCK, HNF1A, HNF1B, HNF4A Young (MODY) 4-gene Panel Maturity-Onset Diabetes of ABCC8, APPL1, BLK, GCK, HNF1A, HNF1B, HNF4A, INS, KCNJ11, 630513 the Young (MODY) Expanded KLF11, NEUROD1, PAX4, PDX1 Genetic Panel Congenital Hyperinsulinism ABCC8, GCK, GLUD1, HADH, HNF1A, HNF4A, KCNJ11, PGM1, PMM2, 630500 Hypoglycemia Genetic Panel SLC16A1, UCP2 Growth and Development Combined Pituitary Hormone GLI1, HESX1, LHX3, LHX4, OTX2, POU1F1, PROKR2, PROP1, -

An Interesting Case of Young Onset Diabetes Mellitus
International Journal of Research in Medical Sciences Gulati S et al. Int J Res Med Sci. 2017 Sep;5(9):4178-4180 www.msjonline.org pISSN 2320-6071 | eISSN 2320-6012 DOI: http://dx.doi.org/10.18203/2320-6012.ijrms20174007 Case Report An interesting case of young onset diabetes mellitus Shipra Gulati*, Tanvi Batra, Akshay A. Dhamne, Vijayashree S. Gokhale Department of Medicine, Dr. DY Patil Medical College and Hospital, Pimpri, Pune, Maharashtra, India Received: 23 June 2017 Accepted: 22 July 2017 *Correspondence: Dr. Shipra Gulati, E-mail: [email protected] Copyright: © the author(s), publisher and licensee Medip Academy. This is an open-access article distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited. ABSTRACT A 24 years old female, was admitted with symptoms of urinary tract infection. She was married and had bad obstetric history. She was known diabetic for 16 years of age and was on regular treatment with injection human insulin mixtard since the time of diagnosis, but had no episode of diabetic ketosis/ ketoacidosis. She had a positive family history of diabetes. She was further evaluated and was found to have normal C peptide levels and islet cell antibodies were found to be negative. Hence, the possibility of MODY (monogenic diabetes) was considered. Her genetic testing could not be done due to financial constraints. But a trial of sulfonylureas was given along with reduction in the dose of insulin to which she responded well and is presently well controlled. -

Servicios GENYCA 20171218 ENGLISH
RARE DISEASES GENYCA_2018 TAT* CODE TEST OMIM ANALYSIS OF: GENES SAMPLE SPECIALITY (turnaraound time) 5 ml blood EDTA/Saliva SRD5A1-S Steroid 5-alpha-reductase *184753 cds SRD5A1 1 month Oncologic on swab 5 ml blood EDTA/Saliva ABL1-S ABL1, Gene *189980 cds ABL1 3 months Oncologic on swab 5 ml blood EDTA/Saliva GCDH-S Glutaric Acidemia Type I 231670 cds GCDH 4 weeks Metabolic on swab ACAT1, BCKDHA, BCKDHB, DBT, 251000, 251100, 251110, GCDH, HMGCL, IVD, MMAA, ACID-N Isolated Methylmalonic Acidemia 251120, 277410, 607481, cds 5 ml blood EDTA 3 months Metabolic MMAB, MMACHC, MCCC1, 607568, 608419, 609058 MCCC2, MUT, PCCA, PCCB 5 ml blood EDTA/Saliva SLC4A1-S Renal Tubular Distal Acidosis 611590 cds SLC4A1 6 weeks Hematologic on swab 5 ml blood EDTA/Saliva AUH-S 3-methylglutaconic aciduria, type I 250950 cds AUH 2 months Metabolic on swab 5 ml blood EDTA/Saliva IDH2-S D-2-hydroxyglutaric aciduria 2 613657 cds IDH2 6 weeks Multisystemic on swab ACYL-COA Dehydrogenase Medium-Chain, c.985A>G 5 ml blood EDTA/Saliva ACADM-V 201450 ACADM 1 month Metabolic Deficiency of (p.Lys304Glu) on swab ACYL-COA Dehydrogenase Medium-Chain, ACADM-S 201450 cds ACADM 5 ml blood EDTA 2 months Metabolic Deficiency of 5 ml blood EDTA/Saliva FGFR3-V Achondroplasia 100800 p.G380R, p.G375C FGFR3 2 weeks Musculoskeletal on swab 5 ml blood EDTA/Saliva ITGB2-S Leukocyte adhesion deficiency (LAD) 116920 cds ITGB2 2 months Hematologic on swab 5 ml blood EDTA/Saliva ADIPOQ-V Adiponectin (Hipoadiponectinemia) 612556 c.276G>T ADIPOQ 1 week Metabolic on swab ABCD1-D Adrenoleukodystrophy -

Genome-Wide Meta-Analysis Identifies Five New Susceptibility Loci for Pancreatic Cancer
Genome-wide meta-analysis identifies five new susceptibility loci for pancreatic cancer The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Klein, A. P., B. M. Wolpin, H. A. Risch, R. Z. Stolzenberg-Solomon, E. Mocci, M. Zhang, F. Canzian, et al. 2018. “Genome-wide meta- analysis identifies five new susceptibility loci for pancreatic cancer.” Nature Communications 9 (1): 556. doi:10.1038/ s41467-018-02942-5. http://dx.doi.org/10.1038/s41467-018-02942-5. Published Version doi:10.1038/s41467-018-02942-5 Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:35015015 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA ARTICLE DOI: 10.1038/s41467-018-02942-5 OPEN Genome-wide meta-analysis identifies five new susceptibility loci for pancreatic cancer Alison P. Klein et al.# In 2020, 146,063 deaths due to pancreatic cancer are estimated to occur in Europe and the United States combined. To identify common susceptibility alleles, we performed the largest pancreatic cancer GWAS to date, including 9040 patients and 12,496 controls of European 1234567890():,; ancestry from the Pancreatic Cancer Cohort Consortium (PanScan) and the Pancreatic Cancer Case-Control Consortium (PanC4). Here, we find significant evidence of a novel association at rs78417682 (7p12/TNS3, P = 4.35 × 10−8). Replication of 10 promising signals in up to 2737 patients and 4752 controls from the PANcreatic Disease ReseArch (PAN- DoRA) consortium yields new genome-wide significant loci: rs13303010 at 1p36.33 (NOC2L, P = 8.36 × 10−14), rs2941471 at 8q21.11 (HNF4G, P = 6.60 × 10−10), rs4795218 at 17q12 (HNF1B, P = 1.32 × 10−8), and rs1517037 at 18q21.32 (GRP, P = 3.28 × 10−8). -

The Mutation Spectrum of Maturity Onset Diabetes of the Young (MODY)-Associated Genes Among Western Siberia Patients
Journal of Personalized Medicine Article The Mutation Spectrum of Maturity Onset Diabetes of the Young (MODY)-Associated Genes among Western Siberia Patients Dinara E. Ivanoshchuk 1,2,* , Elena V. Shakhtshneider 1,2 , Oksana D. Rymar 2, Alla K. Ovsyannikova 2, Svetlana V. Mikhailova 1, Veniamin S. Fishman 1, Emil S. Valeev 1, Pavel S. Orlov 1,2 and Mikhail I. Voevoda 1 1 Federal Research Center Institute of Cytology and Genetics, Siberian Branch of Russian Academy of Sciences (SB RAS), Prospekt Lavrentyeva 10, 630090 Novosibirsk, Russia; [email protected] (E.V.S.); [email protected] (S.V.M.); [email protected] (V.S.F.); [email protected] (E.S.V.); [email protected] (P.S.O.); [email protected] (M.I.V.) 2 Institute of Internal and Preventive Medicine—Branch of Institute of Cytology and Genetics, SB RAS, Bogatkova Str. 175/1, 630004 Novosibirsk, Russia; [email protected] (O.D.R.); [email protected] (A.K.O.) * Correspondence: [email protected] or [email protected]; Tel.: +7-(383)-363-4963; Fax: +7-(383)-333-1278 Abstract: Maturity onset diabetes of the young (MODY) is a congenital form of diabetes characterized by onset at a young age and a primary defect in pancreatic-β-cell function. Currently, 14 subtypes of MODY are known, and each is associated with mutations in a specific gene: HNF4A, GCK, HNF1A, PDX1, HNF1B, NEUROD1, KLF11, CEL, PAX4, INS, BLK, KCNJ11, ABCC8, and APPL1. The most common subtypes of MODY are associated with mutations in the genes GCK, HNF1A, HNF4A, and HNF1B. -

OZGROW DIAGNOSIS CODES H Growth Disorders
OZGROW DIAGNOSIS CODES H Growth Disorders HA Short Stature (unspecified) HAA Endocrine HAAA Hypothyroid (Also code NA. Code JBBBEB if due to secondary hyperprolactinaemia) HAAB Cushings (Also code AABA + MAB). HAAC Genetic GH deficiency HAAD Growth Hormone Deficiency HAADA Isolated GH deficiency HAADAA Congenital idiopathic HAADAAA Partial GH deficiency HAADAAZ Other (Specify) HAADAB Pituitary hypoplasia and aplasia (also code JBAA) HAADAC Radiotherapy HAADACA Cranial > 24 Gy HAADACB Cranial >18Gy <=24Gy HAADACC Craniospinal HAADACZ Other (Specify) HAADAD Trauma HAADAE Malignancy HAADAEA Histiocytosis HAADAEB Leukaemia HAADAEC Glioma HAADAED Germinoma HAADAEE Astrocytoma HAADAEF Ependymona HAADAEG Medullablastoma HAADAEH Nasopharyngeal tumour HAADAEI Lymphoma HAADAEZ Other (Specify) HAADAF Neurosecretory HAADAG CNS malformation HAADAGA Septo optic dysplasia (Also code JAAA) HAADAGB Optic hypoplasia (Also code JAAB) HAADAGC Corpus callosum defect (Also Code JAAC) HAADAGD Midline palatal cleft HAADAGE Empty sella HAADAGZ Other (Specify) HAADAH Craniopharyngioma HAADAZ Other (Specify) 1 HAADB Multiple pituitary deficiencies HAADBA Congenital idiopathic HAADBB Pituitary hypoplasia and aplasia HAADBC Radiotherapy HAADBCA Cranial > 24 Gy HAADBCB Cranial >18Gy <=24Gy HAADBCC Craniospinal HAADBCZ Other (Specify) HAADBD Trauma HAADBE Malignancy HAADBEA Histiocytosis HAADBEB Leukaemia HAADBEC Glioma HAADBED Germinoma HAADBEE Astrocytoma HAADBEF Ependymona HAADBEG Medullablastoma HAADBEH Nasopharyngeal tumour HAADBEI Lymphoma HAADBEZ Other (Specify)