Ryr1 Deficiency in Congenital Myopathies Disrupts Excitation-Contraction Coupling
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

RESEARCH ARTICLE Body Weight-Dependent Troponin T Alternative Splicing Is Evolutionarily Conserved from Insects to Mammals and I
1523 The Journal of Experimental Biology 214, 1523-1532 © 2011. Published by The Company of Biologists Ltd doi:10.1242/jeb.051763 RESEARCH ARTICLE Body weight-dependent troponin T alternative splicing is evolutionarily conserved from insects to mammals and is partially impaired in skeletal muscle of obese rats Rudolf J. Schilder1,*, Scot R. Kimball1, James H. Marden2 and Leonard S. Jefferson1 1Department of Cellular and Molecular Physiology, The Pennsylvania State University College of Medicine, 500 University Drive, Hershey, PA 17033, USA and 2Department of Biology, The Pennsylvania State University, 208 Mueller Lab, University Park, PA 16802, USA *Author for correspondence ([email protected]) Accepted 19 January 2011 SUMMARY Do animals know at a physiological level how much they weigh, and, if so, do they make homeostatic adjustments in response to changes in body weight? Skeletal muscle is a likely tissue for such plasticity, as weight-bearing muscles receive mechanical feedback regarding body weight and consume ATP in order to generate forces sufficient to counteract gravity. Using rats, we examined how variation in body weight affected alternative splicing of fast skeletal muscle troponin T (Tnnt3), a component of the thin filament that regulates the actin–myosin interaction during contraction and modulates force output. In response to normal growth and experimental body weight increases, alternative splicing of Tnnt3 in rat gastrocnemius muscle was adjusted in a quantitative fashion. The response depended on weight per se, as externally attached loads had the same effect as an equal change in actual body weight. Examining the association between Tnnt3 alternative splicing and ATP consumption rate, we found that the Tnnt3 splice form profile had a significant association with nocturnal energy expenditure, independently of effects of weight. -

Bit.Bio Ioskeletal Myocytes Faqs
ioSkeletal Myocytes Early Access Product Catalogue no: EA1200 Frequently Asked Questions Doc no: NPI-0002-FAQ V-01 For research use only bit.bio The Dorothy Hodgkin Building Babraham Research Campus Cambridge CB22 3FH United Kingdom www.bit.bio Customer support: [email protected] [email protected] ioSkeletal Myocytes Questions adressed Early Access Product Catalogue no: EA1200 Shipping, ordering and delivery Frequently Asked Questions 1. What format will the cells be delivered to clients: Doc no: NPI-0002-FAQ V-01 frozen vials or pre-plated cells? 2. How can I contact you if I have a question? For research use only bit.bio Cell revival and experiments The Dorothy Hodgkin Building Babraham Research Campus 1. Are ioSkeletal Myocytes cells fully differentiated? Cambridge CB22 3FH 2. Can you propagate ioSkeletal Myocytes cells once received? United Kingdom 3. What seeding density do you recommend for the ioSkeletal Myocytes? www.bit.bio 4. How are cells cultivated? 5. How soon after delivery can ioSkeletal Myocytes be used for Customer support: [email protected] experiments? [email protected] Product information & quality control 1. Why is opti-ox better than other methods of cellular reprogramming? 2. What were the cells of origin for ioSkeletal Myocytes? 3. Do you have donor consent for the parental hiPSCs? 4. Do you use viral vectors to manufacture ioSkeletal Myocytes? 5. What is the host and transgene used to generate cells? 6. What substances other than KnockOut serum may be present in the freezing medium? 7. What quality control is performed on the ioSkeletal Myocytes? 8. How does bit.bio confirm its cell lines are free from contamination? 9. -

Nuclear Envelope Laminopathies: Evidence for Developmentally Inappropriate Nuclear Envelope-Chromatin Associations
Nuclear Envelope Laminopathies: Evidence for Developmentally Inappropriate Nuclear Envelope-Chromatin Associations by Jelena Perovanovic M.S. in Molecular Biology and Physiology, September 2009, University of Belgrade M.Phil. in Molecular Medicine, August 2013, The George Washington University A Dissertation submitted to The Faculty of The Columbian College of Arts and Sciences of The George Washington University in partial fulfillment of the requirements for the degree of Doctor of Philosophy August 31, 2015 Dissertation directed by Eric P. Hoffman Professor of Integrative Systems Biology The Columbian College of Arts and Sciences of The George Washington University certifies that Jelena Perovanovic has passed the Final Examination for the degree of Doctor of Philosophy as of May 5, 2015. This is the final and approved form of the dissertation. Nuclear Envelope Laminopathies: Evidence for Developmentally Inappropriate Nuclear Envelope-Chromatin Associations Jelena Perovanovic Dissertation Research Committee: Eric P. Hoffman, Professor of Integrative Systems Biology, Dissertation Director Anamaris Colberg-Poley, Professor of Integrative Systems Biology, Committee Member Robert J. Freishtat, Associate Professor of Pediatrics, Committee Member Vittorio Sartorelli, Senior Investigator, National Institutes of Health, Committee Member ii © Copyright 2015 by Jelena Perovanovic All rights reserved iii Acknowledgments I am deeply indebted to countless individuals for their support and encouragement during the past five years of graduate studies. First and foremost, I would like to express my gratitude to my mentor, Dr. Eric P. Hoffman, for his unwavering support and guidance, and keen attention to my professional development. This Dissertation would not have been possible without the critical input he provided and the engaging environment he created. -

Troponin Variants in Congenital Myopathies: How They Affect Skeletal Muscle Mechanics
International Journal of Molecular Sciences Review Troponin Variants in Congenital Myopathies: How They Affect Skeletal Muscle Mechanics Martijn van de Locht , Tamara C. Borsboom, Josine M. Winter and Coen A. C. Ottenheijm * Department of Physiology, Amsterdam Cardiovascular Sciences, Amsterdam UMC, Location VUmc, 1081 HZ Amsterdam, The Netherlands; [email protected] (M.v.d.L.); [email protected] (T.C.B.); [email protected] (J.M.W.) * Correspondence: [email protected]; Tel.: +31-(0)-20-444-8123 Abstract: The troponin complex is a key regulator of muscle contraction. Multiple variants in skeletal troponin encoding genes result in congenital myopathies. TNNC2 has been implicated in a novel congenital myopathy, TNNI2 and TNNT3 in distal arthrogryposis (DA), and TNNT1 and TNNT3 in nemaline myopathy (NEM). Variants in skeletal troponin encoding genes compromise sarcomere function, e.g., by altering the Ca2+ sensitivity of force or by inducing atrophy. Several potential therapeutic strategies are available to counter the effects of variants, such as troponin activators, introduction of wild-type protein through AAV gene therapy, and myosin modulation to improve muscle contraction. The mechanisms underlying the pathophysiological effects of the variants in skeletal troponin encoding genes are incompletely understood. Furthermore, limited knowledge is available on the structure of skeletal troponin. This review focusses on the physiology of slow and fast skeletal troponin and the pathophysiology of reported variants in skeletal troponin encoding genes. A better understanding of the pathophysiological effects of these variants, together with enhanced knowledge regarding the structure of slow and fast skeletal troponin, will direct the development of Citation: van de Locht, M.; treatment strategies. -

Analysis of the Dystrophin Interactome
Analysis of the dystrophin interactome Dissertation In fulfillment of the requirements for the degree “Doctor rerum naturalium (Dr. rer. nat.)” integrated in the International Graduate School for Myology MyoGrad in the Department for Biology, Chemistry and Pharmacy at the Freie Universität Berlin in Cotutelle Agreement with the Ecole Doctorale 515 “Complexité du Vivant” at the Université Pierre et Marie Curie Paris Submitted by Matthew Thorley born in Scunthorpe, United Kingdom Berlin, 2016 Supervisor: Simone Spuler Second examiner: Sigmar Stricker Date of defense: 7th December 2016 Dedicated to My mother, Joy Thorley My father, David Thorley My sister, Alexandra Thorley My fiancée, Vera Sakhno-Cortesi Acknowledgements First and foremost, I would like to thank my supervisors William Duddy and Stephanie Duguez who gave me this research opportunity. Through their combined knowledge of computational and practical expertise within the field and constant availability for any and all assistance I required, have made the research possible. Their overarching support, approachability and upbeat nature throughout, while granting me freedom have made this year project very enjoyable. The additional guidance and supported offered by Matthias Selbach and his team whenever required along with a constant welcoming invitation within their lab has been greatly appreciated. I thank MyoGrad for the collaboration established between UPMC and Freie University, creating the collaboration within this research project possible, and offering research experience in both the Institute of Myology in Paris and the Max Delbruck Centre in Berlin. Vital to this process have been Gisele Bonne, Heike Pascal, Lidia Dolle and Susanne Wissler who have aided in the often complex processes that I am still not sure I fully understand. -
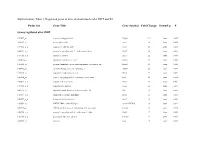
Probe Set Gene Title Gene Symbol Fold Change Down/Up P
Supplementary Table 1. Regulated genes in mice skeletal muscle after DHT and E2 Probe Set Gene Title Gene Symbol Fold Change Down/Up P Genes regulated after DHT 1419487_at myosin binding protein H Mybph 11.9 down 0.000 1415927_at Actin. alpha. cardiac Actc1 3.8 down 0.000 1419606_a_at troponin T1. skeletal. slow Tnnt1 2.8 down 0.004 1448553_at myosin. heavy polypeptide 7. cardiac muscle. beta Myh7 2.6 down 0.001 1417765_a_at amylase 1. salivary Amy1 2.5 down 0.000 1416411_at glutathione S-transferase. mu 2 Gstm2 2.5 down 0.000 1418492_at gremlin 2 homolog. cysteine knot superfamily (Xenopus laevis) Grem2 2.4 down 0.000 1424392_at alcohol dehydrogenase. iron containing. 1 Adhfe1 2.2 down 0.000 1418370_at troponin C. cardiac/slow skeletal Tnnc1 2.2 down 0.002 1448394_at myosin. light polypeptide 2. regulatory. cardiac. slow Myl2 2.2 down 0.004 1450813_a_at troponin I. skeletal. slow 1 Tnni1 2.1 down 0.001 1449996_a_at tropomyosin 3. gamma Tpm3 2.1 down 0.017 1449118_at dihydrolipoamide branched chain transacylase E2 Dbt 1.8 down 0.002 1418507_s_at suppressor of cytokine signaling 2 Socs2 1.8 down 0.038 1449059_a_at 3-oxoacid CoA transferase 1 Oxct1 1.8 down 0.000 1453552_at RIKEN cDNA 2310014F07 gene 2310014F07Rik 1.8 down 0.012 1436913_at CDC14 cell division cycle 14 homolog A (S. cerevisiae) Cdc14a 1.8 down 0.014 1448554_s_at myosin. heavy polypeptide 6. cardiac muscle. alpha Myh6 1.7 down 0.008 1427168_a_at procollagen. type XIV. alpha 1 Col14a1 1.7 down 0.005 1423607_at lumican Lum 1.7 down 0.005 1451310_a_at cathepsin L Ctsl 1.7 down 0.001 1452363_a_at ATPase. -

Protein Aggregate Myopathies
Review Article Protein aggregate myopathies M. C. Sharma, H. H. Goebel1 All India Institute of Medical Sciences, New Delhi, India and 1Department of Neuropathology, Johannes Gutenberg University Medical Center, Mainz, Germany Protein aggregate myopathies (PAM) are an emerging group Aggegation of proteins within muscle cells may be of lyso- of muscle diseases characterized by structural abnormali- somal or nonlysosomal nature. The former occurs, for instance, ties. Protein aggregate myopathies are marked by the ag- in the neuronal ceroid-lipofuscinoses (NCL) as components gregation of intrinsic proteins within muscle fibers and fall of lipopigments, their accretion being a morphological hall- into four major groups or conditions: (1) desmin-related mark in the NCL.[1] This lysosomal accumulation of myopathies (DRM) that include desminopathies, a-B lipopigments renders the NCL a member of the lysosomal dis- crystallinopathies, selenoproteinopathies caused by muta- orders among which type-II glycogenosis, Niemann-Pick dis- tions in the, a-B crystallin and selenoprotein N1 genes, (2) ease, mucolipidosis IV and others may affect the skeletal hereditary inclusion body myopathies, several of which have muscle. been linked to different chromosomal gene loci, but with as Nonlysosomal accumulation of proteins within muscle fibers yet unidentified protein product, (3) actinopathies marked is a hallmark of a recently emerging subgroup of myopathies, by mutations in the sarcomeric ACTA1 gene, and (4) called protein aggregate myopathies (PAM). The cause -

Supplementary Table 5A
CE2/+ f/f CA/+ CE2/+ f/f Supplementary Table 5A: Pathway enrichment analysis for differentially expressed genes between Nkx3.1 ; Pten ; Braf and Nkx3.1 ; Pten mouse prostate tumors Pathway Gene Number Enrichment Normalized p-value False Family-wise Rank Leading Edge Genes Set of genes Score enrichment Discovery Error Rate at Details in the score Rate p-value Max pathway q-value TPM2 ACTG2 MYL9 ACTA2 VCL DES MYLK ITGA1 Details TPM4 SORBS1 CALM2 ITGB5 PXN LMOD1 TNNC2 1 REACTOME_MUSCLE_CONTRACTION ... 30 0.78 2.58 0 0 0 2180 MYL1 MYH11 Details TPM2 ACTG2 MYL9 ACTA2 VCL MYLK ITGA1 2 REACTOME_SMOOTH_MUSCLE_CONTRACTION ... 20 0.81 2.39 0 0 0 1609 TPM4 SORBS1 CALM2 ITGB5 PXN LMOD1 MYH11 ITGA3 LAMB3 ITGB4 THBS1 LAMA3 CD44 ITGA6 ITGAV ITGB1 SPP1 LAMC2 ITGA1 SDC1 RELN LAMC1 FN1 ITGB5 THBS2 COL1A2 ITGA9 ITGB6 SDC2 HSPG2 TNC COL6A1 COL4A2 COL6A2 Details COL4A6 ITGA5 COL4A1 LAMA1SV2B COL5A1 3 KEGG_ECM_RECEPTOR_INTERACTION ... 59 0.63 2.32 0 0 0 3812 COL6A3 COL3A1 ITGA11 COL5A2 ITGA10 Details ACTIN1 FLNC FBLIM1 ITGB1 FLNA RSU1 FERMT2 4 REACTOME_CELLEXTRACELLULAR_MATRIX_INTERACTIONS ... 13 0.81 2.16 0 0 0.001 1193 PXN ILK ITGA3 ITGB4 THBS1 ITGA6 ITGAV ITGB1 SPP1 ITGA1 LAMC1 FN1 ITGB5 COL1A2 CDH1 BCRAR1 ITGA9 ITGB6 COL4A5 TNC COL4A2 ITGA5 COL4A1 Details LAMA1 PTPN1 FGG PECAM1 FGA FBN1 ITGA11 5 REACTOME_INTEGRIN_CELL_SURFACE_INTERACTIONS ... 62 0.57 2.09 0 0 0.002 3812 ITGA10 COL1A1 LAMA2 LAMB2 ITGA3 CCND1 LAMB3 ITGB4 THBS1 FLNB LAMA3 ACTN1 IGF1R CCND2 ITGA6 ITGAV FLNC MYL9 CAV1 ITGB1 VEGF1 PDGFRA SPP1 FLNA MAP2K1 PDGFC VCL PPP1CC MYLK LAMC2 CAPN2 CDC42 ITGA1 RELN LAMC1 AKT3 FN1 CAV2 PDGFRB ITGB5 PXN ILK PDGFB THBS2 DOCK1 COL1A2 FLT1 FIGF BCAR1 ITGA9 ITGB6 PAK3 TNC COL6A1 COL4A2 COL6A2 COL4A6 ITGA5 COL4A1 LAMA1 Details BCL2 COL5A1 PGF ZYX COL6A3 COL3A1 ITGA11 6 KEGG_FOCAL_ADHESION .. -

Supplemental Table 1
Supplemental Data Supplemental Table 1. Genes differentially regulated by Ad-KLF2 vs. Ad-GFP infected EC. Three independent genome-wide transcriptional profiling experiments were performed, and significantly regulated genes were identified. Color-coding scheme: Up, p < 1e-15 Up, 1e-15 < p < 5e-10 Up, 5e-10 < p < 5e-5 Up, 5e-5 < p <.05 Down, p < 1e-15 As determined by Zpool Down, 1e-15 < p < 5e-10 Down, 5e-10 < p < 5e-5 Down, 5e-5 < p <.05 p<.05 as determined by Iterative Standard Deviation Algorithm as described in Supplemental Methods Ratio RefSeq Number Gene Name 1,058.52 KRT13 - keratin 13 565.72 NM_007117.1 TRH - thyrotropin-releasing hormone 244.04 NM_001878.2 CRABP2 - cellular retinoic acid binding protein 2 118.90 NM_013279.1 C11orf9 - chromosome 11 open reading frame 9 109.68 NM_000517.3 HBA2;HBA1 - hemoglobin, alpha 2;hemoglobin, alpha 1 102.04 NM_001823.3 CKB - creatine kinase, brain 96.23 LYNX1 95.53 NM_002514.2 NOV - nephroblastoma overexpressed gene 75.82 CeleraFN113625 FLJ45224;PTGDS - FLJ45224 protein;prostaglandin D2 synthase 21kDa 74.73 NM_000954.5 (brain) 68.53 NM_205545.1 UNQ430 - RGTR430 66.89 NM_005980.2 S100P - S100 calcium binding protein P 64.39 NM_153370.1 PI16 - protease inhibitor 16 58.24 NM_031918.1 KLF16 - Kruppel-like factor 16 46.45 NM_024409.1 NPPC - natriuretic peptide precursor C 45.48 NM_032470.2 TNXB - tenascin XB 34.92 NM_001264.2 CDSN - corneodesmosin 33.86 NM_017671.3 C20orf42 - chromosome 20 open reading frame 42 33.76 NM_024829.3 FLJ22662 - hypothetical protein FLJ22662 32.10 NM_003283.3 TNNT1 - troponin T1, skeletal, slow LOC388888 (LOC388888), mRNA according to UniGene - potential 31.45 AK095686.1 CONFLICT - LOC388888 (na) according to LocusLink. -

LOXL2-Mediated H3K4 Oxidation Reduces Chromatin Accessibility in Triple-Negative Breast Cancer Cells
Oncogene (2020) 39:79–121 https://doi.org/10.1038/s41388-019-0969-1 ARTICLE LOXL2-mediated H3K4 oxidation reduces chromatin accessibility in triple-negative breast cancer cells 1 1 2 1 1 1 J. P. Cebrià-Costa ● L. Pascual-Reguant ● A. Gonzalez-Perez ● G. Serra-Bardenys ● J. Querol ● M. Cosín ● 1,3 4 4 2 5 6 G. Verde ● R. A. Cigliano ● W. Sanseverino ● S. Segura-Bayona ● A. Iturbide ● D. Andreu ● 1 1,7 1 1,7,8,9 10 6,10 P. Nuciforo ● C. Bernado-Morales ● V. Rodilla ● J. Arribas ● J. Yelamos ● A. Garcia de Herreros ● 2 1 T. H. Stracker ● S. Peiró Received: 28 January 2019 / Revised: 8 July 2019 / Accepted: 9 August 2019 / Published online: 28 August 2019 © The Author(s) 2019. This article is published with open access Abstract Oxidation of H3 at lysine 4 (H3K4ox) by lysyl oxidase-like 2 (LOXL2) generates an H3 modification with an unknown physiological function. We find that LOXL2 and H3K4ox are higher in triple-negative breast cancer (TNBC) cell lines and patient-derived xenografts (PDXs) than those from other breast cancer subtypes. ChIP-seq revealed that H3K4ox is located primarily in heterochromatin, where it is involved in chromatin compaction. Knocking down LOXL2 reduces H3K4ox levels 1234567890();,: 1234567890();,: and causes chromatin decompaction, resulting in a sustained activation of the DNA damage response (DDR) and increased susceptibility to anticancer agents. This critical role that LOXL2 and oxidized H3 play in chromatin compaction and DDR suggests that functionally targeting LOXL2 could be a way to sensitize TNBC cells to conventional therapy. -

Transcriptional Profiling Reveals Extraordinary Diversity Among
TOOLS AND RESOURCES Transcriptional profiling reveals extraordinary diversity among skeletal muscle tissues Erin E Terry1, Xiping Zhang2, Christy Hoffmann1, Laura D Hughes3, Scott A Lewis1, Jiajia Li1, Matthew J Wallace1, Lance A Riley2, Collin M Douglas2, Miguel A Gutierrez-Monreal2, Nicholas F Lahens4, Ming C Gong5, Francisco Andrade5, Karyn A Esser2, Michael E Hughes1* 1Division of Pulmonary and Critical Care Medicine, Washington University School of Medicine, St. Louis, United States; 2Department of Physiology and Functional Genomics, University of Florida College of Medicine, Gainesville, United States; 3Department of Integrative, Structural and Computational Biology, The Scripps Research Institute, La Jolla, United States; 4Institute for Translational Medicine and Therapeutics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, United States; 5Department of Physiology, University of Kentucky School of Medicine, Lexington, United States Abstract Skeletal muscle comprises a family of diverse tissues with highly specialized functions. Many acquired diseases, including HIV and COPD, affect specific muscles while sparing others. Even monogenic muscular dystrophies selectively affect certain muscle groups. These observations suggest that factors intrinsic to muscle tissues influence their resistance to disease. Nevertheless, most studies have not addressed transcriptional diversity among skeletal muscles. Here we use RNAseq to profile mRNA expression in skeletal, smooth, and cardiac muscle tissues from mice and rats. Our data set, MuscleDB, reveals extensive transcriptional diversity, with greater than 50% of transcripts differentially expressed among skeletal muscle tissues. We detect mRNA expression of *For correspondence: hundreds of putative myokines that may underlie the endocrine functions of skeletal muscle. We [email protected] identify candidate genes that may drive tissue specialization, including Smarca4, Vegfa, and Competing interests: The Myostatin. -

Altered Gene Expression in Circulating Immune Cells Following a 24-Hour Passive Dehydration
University of Connecticut OpenCommons@UConn Master's Theses University of Connecticut Graduate School 5-10-2020 Altered Gene Expression in Circulating Immune Cells Following a 24-Hour Passive Dehydration Aidan Fiol [email protected] Follow this and additional works at: https://opencommons.uconn.edu/gs_theses Recommended Citation Fiol, Aidan, "Altered Gene Expression in Circulating Immune Cells Following a 24-Hour Passive Dehydration" (2020). Master's Theses. 1480. https://opencommons.uconn.edu/gs_theses/1480 This work is brought to you for free and open access by the University of Connecticut Graduate School at OpenCommons@UConn. It has been accepted for inclusion in Master's Theses by an authorized administrator of OpenCommons@UConn. For more information, please contact [email protected]. Altered Gene Expression in Circulating Immune Cells Following a 24- Hour Passive Dehydration Aidan Fiol B.S., University of Connecticut, 2018 A Thesis Submitted in Partial Fulfillment of the Requirements for the Degree of Master of Science At the University of Connecticut 2020 i copyright by Aidan Fiol 2020 ii APPROVAL PAGE Master of Science Thesis Altered Gene Expression in Circulating Immune Cells Following a 24- Hour Passive Dehydration Presented by Aidan Fiol, B.S. Major Advisor__________________________________________________________________ Elaine Choung-Hee Lee, Ph.D. Associate Advisor_______________________________________________________________ Douglas J. Casa, Ph.D. Associate Advisor_______________________________________________________________ Robert A. Huggins, Ph.D. University of Connecticut 2020 iii ACKNOWLEDGEMENTS I’d like to thank my committee members. Dr. Lee, you have been an amazing advisor, mentor and friend to me these past few years. Your advice, whether it was how to be a better writer or scientist, or just general life advice given on our way to get coffee, has helped me grow as a researcher and as a person.