Analysis of the Dystrophin Interactome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Data
Figure 2S 4 7 A - C 080125 CSCs 080418 CSCs - + IFN-a 48 h + IFN-a 48 h + IFN-a 72 h 6 + IFN-a 72 h 3 5 MRFI 4 2 3 2 1 1 0 0 MHC I MHC II MICA MICB ULBP-1 ULBP-2 ULBP-3 ULBP-4 MHC I MHC II MICA MICB ULBP-1 ULBP-2 ULBP-3 ULBP-4 7 B 13 080125 FBS - D 080418 FBS - + IFN-a 48 h 12 + IFN-a 48 h + IFN-a 72 h + IFN-a 72 h 6 080125 FBS 11 10 5 9 8 4 7 6 3 MRFI 5 4 2 3 2 1 1 0 0 MHC I MHC II MICA MICB ULBP-1 ULBP-2 ULBP-3 ULBP-4 MHC I MHC II MICA MICB ULBP-1 ULBP-2 ULBP-3 ULBP-4 Molecule Molecule FIGURE 4S FIGURE 5S Panel A Panel B FIGURE 6S A B C D Supplemental Results Table 1S. Modulation by IFN-α of APM in GBM CSC and FBS tumor cell lines. Molecule * Cell line IFN-α‡ HLA β2-m# HLA LMP TAP1 TAP2 class II A A HC§ 2 7 10 080125 CSCs - 1∞ (1) 3 (65) 2 (91) 1 (2) 6 (47) 2 (61) 1 (3) 1 (2) 1 (3) + 2 (81) 11 (80) 13 (99) 1 (3) 8 (88) 4 (91) 1 (2) 1 (3) 2 (68) 080125 FBS - 2 (81) 4 (63) 4 (83) 1 (3) 6 (80) 3 (67) 2 (86) 1 (3) 2 (75) + 2 (99) 14 (90) 7 (97) 5 (75) 7 (100) 6 (98) 2 (90) 1 (4) 3 (87) 080418 CSCs - 2 (51) 1 (1) 1 (3) 2 (47) 2 (83) 2 (54) 1 (4) 1 (2) 1 (3) + 2 (81) 3 (76) 5 (75) 2 (50) 2 (83) 3 (71) 1 (3) 2 (87) 1 (2) 080418 FBS - 1 (3) 3 (70) 2 (88) 1 (4) 3 (87) 2 (76) 1 (3) 1 (3) 1 (2) + 2 (78) 7 (98) 5 (99) 2 (94) 5 (100) 3 (100) 1 (4) 2 (100) 1 (2) 070104 CSCs - 1 (2) 1 (3) 1 (3) 2 (78) 1 (3) 1 (2) 1 (3) 1 (3) 1 (2) + 2 (98) 8 (100) 10 (88) 4 (89) 3 (98) 3 (94) 1 (4) 2 (86) 2 (79) * expression of APM molecules was evaluated by intracellular staining and cytofluorimetric analysis; ‡ cells were treatead or not (+/-) for 72 h with 1000 IU/ml of IFN-α; # β-2 microglobulin; § β-2 microglobulin-free HLA-A heavy chain; ∞ values are indicated as ratio between the mean of fluorescence intensity of cells stained with the selected mAb and that of the negative control; bold values indicate significant MRFI (≥ 2). -

Supplemental Information to Mammadova-Bach Et Al., “Laminin Α1 Orchestrates VEGFA Functions in the Ecosystem of Colorectal Carcinogenesis”
Supplemental information to Mammadova-Bach et al., “Laminin α1 orchestrates VEGFA functions in the ecosystem of colorectal carcinogenesis” Supplemental material and methods Cloning of the villin-LMα1 vector The plasmid pBS-villin-promoter containing the 3.5 Kb of the murine villin promoter, the first non coding exon, 5.5 kb of the first intron and 15 nucleotides of the second villin exon, was generated by S. Robine (Institut Curie, Paris, France). The EcoRI site in the multi cloning site was destroyed by fill in ligation with T4 polymerase according to the manufacturer`s instructions (New England Biolabs, Ozyme, Saint Quentin en Yvelines, France). Site directed mutagenesis (GeneEditor in vitro Site-Directed Mutagenesis system, Promega, Charbonnières-les-Bains, France) was then used to introduce a BsiWI site before the start codon of the villin coding sequence using the 5’ phosphorylated primer: 5’CCTTCTCCTCTAGGCTCGCGTACGATGACGTCGGACTTGCGG3’. A double strand annealed oligonucleotide, 5’GGCCGGACGCGTGAATTCGTCGACGC3’ and 5’GGCCGCGTCGACGAATTCACGC GTCC3’ containing restriction site for MluI, EcoRI and SalI were inserted in the NotI site (present in the multi cloning site), generating the plasmid pBS-villin-promoter-MES. The SV40 polyA region of the pEGFP plasmid (Clontech, Ozyme, Saint Quentin Yvelines, France) was amplified by PCR using primers 5’GGCGCCTCTAGATCATAATCAGCCATA3’ and 5’GGCGCCCTTAAGATACATTGATGAGTT3’ before subcloning into the pGEMTeasy vector (Promega, Charbonnières-les-Bains, France). After EcoRI digestion, the SV40 polyA fragment was purified with the NucleoSpin Extract II kit (Machery-Nagel, Hoerdt, France) and then subcloned into the EcoRI site of the plasmid pBS-villin-promoter-MES. Site directed mutagenesis was used to introduce a BsiWI site (5’ phosphorylated AGCGCAGGGAGCGGCGGCCGTACGATGCGCGGCAGCGGCACG3’) before the initiation codon and a MluI site (5’ phosphorylated 1 CCCGGGCCTGAGCCCTAAACGCGTGCCAGCCTCTGCCCTTGG3’) after the stop codon in the full length cDNA coding for the mouse LMα1 in the pCIS vector (kindly provided by P. -

PARSANA-DISSERTATION-2020.Pdf
DECIPHERING TRANSCRIPTIONAL PATTERNS OF GENE REGULATION: A COMPUTATIONAL APPROACH by Princy Parsana A dissertation submitted to The Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland July, 2020 © 2020 Princy Parsana All rights reserved Abstract With rapid advancements in sequencing technology, we now have the ability to sequence the entire human genome, and to quantify expression of tens of thousands of genes from hundreds of individuals. This provides an extraordinary opportunity to learn phenotype relevant genomic patterns that can improve our understanding of molecular and cellular processes underlying a trait. The high dimensional nature of genomic data presents a range of computational and statistical challenges. This dissertation presents a compilation of projects that were driven by the motivation to efficiently capture gene regulatory patterns in the human transcriptome, while addressing statistical and computational challenges that accompany this data. We attempt to address two major difficulties in this domain: a) artifacts and noise in transcriptomic data, andb) limited statistical power. First, we present our work on investigating the effect of artifactual variation in gene expression data and its impact on trans-eQTL discovery. Here we performed an in-depth analysis of diverse pre-recorded covariates and latent confounders to understand their contribution to heterogeneity in gene expression measurements. Next, we discovered 673 trans-eQTLs across 16 human tissues using v6 data from the Genotype Tissue Expression (GTEx) project. Finally, we characterized two trait-associated trans-eQTLs; one in Skeletal Muscle and another in Thyroid. Second, we present a principal component based residualization method to correct gene expression measurements prior to reconstruction of co-expression networks. -

A Network-Informed Analysis of SARS-Cov-2 and Hemophagocytic Lymphohistiocytosis Genes' Interactions Points to Neutrophil Extr
medRxiv preprint doi: https://doi.org/10.1101/2020.07.01.20144121; this version posted July 2, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license . 1 A network-informed analysis of SARS-CoV-2 and hemophagocytic 2 lymphohistiocytosis genes’ interactions points to Neutrophil Extracellular Traps as 3 mediators of thromBosis in COVID-19 4 5 Jun Ding1, David Earl Hostallero2, Mohamed Reda El Khili2, Gregory Fonseca3, Simon 6 Millette4, Nuzha Noorah3, Myriam Guay-Belzile3, Jonathan Spicer5, Noriko Daneshtalab6, 7 Martin Sirois7, Karine Tremblay8, Amin Emad2,* and Simon Rousseau3,* 8 9 10 1Computational Biology Department, Carnegie Mellon University, Pittsburgh, PA, USA, 15204 11 12 2Department of Electrical and Computer Engineering, McGill University, Montreal, QC, Canada. 13 14 3The Meakins-Christie Laboratories at the Research Institute of the McGill University Heath 15 Centre Research Institute, 1001 Boul. Décarie, Montréal, H4A 3J1, Canada. 16 17 4Goodman Cancer Research Centre, McGill University 18 19 5Division of Thoracic and Upper Gastrointestinal Surgery, McGill University Health Centre 20 Research Institute, 1001 Boul. Décarie, Montréal, H4A 3J1, Canada. 21 22 6School of Pharmacy, Memorial University of Newfoundland, 300 Prince Philip Drive, Health 23 Sciences Center, St. John’s, Newfoundland, Canada, A1B 3V6 24 25 7Montreal Heart Institute and Department of pharmacology and physiology, Faculty of medicine, 26 Université de Montréal. 27 28 8Pharmacology-physiology Department, Faculty of Medicine and Health Sciences, Université de 29 Sherbrooke, Centre intégré universitaire de santé et de services sociaux du Saguenay–Lac-Saint- 30 Jean (Chicoutimi University Hospital) Research Center, Saguenay, QC, Canada. -

Genetic Mutations and Mechanisms in Dilated Cardiomyopathy
Genetic mutations and mechanisms in dilated cardiomyopathy Elizabeth M. McNally, … , Jessica R. Golbus, Megan J. Puckelwartz J Clin Invest. 2013;123(1):19-26. https://doi.org/10.1172/JCI62862. Review Series Genetic mutations account for a significant percentage of cardiomyopathies, which are a leading cause of congestive heart failure. In hypertrophic cardiomyopathy (HCM), cardiac output is limited by the thickened myocardium through impaired filling and outflow. Mutations in the genes encoding the thick filament components myosin heavy chain and myosin binding protein C (MYH7 and MYBPC3) together explain 75% of inherited HCMs, leading to the observation that HCM is a disease of the sarcomere. Many mutations are “private” or rare variants, often unique to families. In contrast, dilated cardiomyopathy (DCM) is far more genetically heterogeneous, with mutations in genes encoding cytoskeletal, nucleoskeletal, mitochondrial, and calcium-handling proteins. DCM is characterized by enlarged ventricular dimensions and impaired systolic and diastolic function. Private mutations account for most DCMs, with few hotspots or recurring mutations. More than 50 single genes are linked to inherited DCM, including many genes that also link to HCM. Relatively few clinical clues guide the diagnosis of inherited DCM, but emerging evidence supports the use of genetic testing to identify those patients at risk for faster disease progression, congestive heart failure, and arrhythmia. Find the latest version: https://jci.me/62862/pdf Review series Genetic mutations and mechanisms in dilated cardiomyopathy Elizabeth M. McNally, Jessica R. Golbus, and Megan J. Puckelwartz Department of Human Genetics, University of Chicago, Chicago, Illinois, USA. Genetic mutations account for a significant percentage of cardiomyopathies, which are a leading cause of conges- tive heart failure. -

New Approaches to Functional Process Discovery in HPV 16-Associated Cervical Cancer Cells by Gene Ontology
Cancer Research and Treatment 2003;35(4):304-313 New Approaches to Functional Process Discovery in HPV 16-Associated Cervical Cancer Cells by Gene Ontology Yong-Wan Kim, Ph.D.1, Min-Je Suh, M.S.1, Jin-Sik Bae, M.S.1, Su Mi Bae, M.S.1, Joo Hee Yoon, M.D.2, Soo Young Hur, M.D.2, Jae Hoon Kim, M.D.2, Duck Young Ro, M.D.2, Joon Mo Lee, M.D.2, Sung Eun Namkoong, M.D.2, Chong Kook Kim, Ph.D.3 and Woong Shick Ahn, M.D.2 1Catholic Research Institutes of Medical Science, 2Department of Obstetrics and Gynecology, College of Medicine, The Catholic University of Korea, Seoul; 3College of Pharmacy, Seoul National University, Seoul, Korea Purpose: This study utilized both mRNA differential significant genes of unknown function affected by the display and the Gene Ontology (GO) analysis to char- HPV-16-derived pathway. The GO analysis suggested that acterize the multiple interactions of a number of genes the cervical cancer cells underwent repression of the with gene expression profiles involved in the HPV-16- cancer-specific cell adhesive properties. Also, genes induced cervical carcinogenesis. belonging to DNA metabolism, such as DNA repair and Materials and Methods: mRNA differential displays, replication, were strongly down-regulated, whereas sig- with HPV-16 positive cervical cancer cell line (SiHa), and nificant increases were shown in the protein degradation normal human keratinocyte cell line (HaCaT) as a con- and synthesis. trol, were used. Each human gene has several biological Conclusion: The GO analysis can overcome the com- functions in the Gene Ontology; therefore, several func- plexity of the gene expression profile of the HPV-16- tions of each gene were chosen to establish a powerful associated pathway, identify several cancer-specific cel- cervical carcinogenesis pathway. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Receptor-Arrestin Interactions: the GPCR Perspective
biomolecules Review Receptor-Arrestin Interactions: The GPCR Perspective Mohammad Seyedabadi 1,2 , Mehdi Gharghabi 3, Eugenia V. Gurevich 4 and Vsevolod V. Gurevich 4,* 1 Department of Toxicology & Pharmacology, Faculty of Pharmacy, Mazandaran University of Medical Sciences, Sari 48471-93698, Iran; [email protected] 2 Pharmaceutical Sciences Research Center, Faculty of Pharmacy, Mazandaran University of Medical Sciences, Sari 48167-75952, Iran 3 Department of Cancer Biology and Genetics, The Ohio State University Wexner Medical Center, Columbus, OH 43210, USA; [email protected] 4 Department of Pharmacology, Vanderbilt University, Nashville, TN 37232, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-615-322-7070; Fax: +1-615-343-6532 Abstract: Arrestins are a small family of four proteins in most vertebrates that bind hundreds of different G protein-coupled receptors (GPCRs). Arrestin binding to a GPCR has at least three functions: precluding further receptor coupling to G proteins, facilitating receptor internalization, and initiating distinct arrestin-mediated signaling. The molecular mechanism of arrestin–GPCR interactions has been extensively studied and discussed from the “arrestin perspective”, focusing on the roles of arrestin elements in receptor binding. Here, we discuss this phenomenon from the “receptor perspective”, focusing on the receptor elements involved in arrestin binding and empha- sizing existing gaps in our knowledge that need to be filled. It is vitally important to understand the role of receptor elements in arrestin activation and how the interaction of each of these elements with arrestin contributes to the latter’s transition to the high-affinity binding state. A more precise knowledge of the molecular mechanisms of arrestin activation is needed to enable the construction of arrestin mutants with desired functional characteristics. -

Integrative Analyses Identify Potential Key Genes and Pathways in Keshan
medRxiv preprint doi: https://doi.org/10.1101/2021.03.12.21253491; this version posted March 15, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Integrative analyses identify potential key genes and pathways in Keshan disease using whole-exome sequencing Jichang Huang1#, Chenqing Zheng2#, Rong Luo1#, Mingjiang Liu3, Qingquan Gu4, Jinshu Li5, Xiushan Wu6, Zhenglin Yang3, Xia Shen2*, Xiaoping Li3* 1 Institute of Geriatric Cardiovascular Disease, Chengdu Medical College, Chengdu, People’s Republic of China 2 State Key Laboratory of Biocontrol, School of Life Sciences, Sun Yat-sen University, Guangzhou, China 3 Department of Cardiology, Hospital of the University of Electronic Science and Technology of China and Sichuan Provincial People’s Hospital, Chengdu, Sichuan, China 4 Shenzhen RealOmics (Biotech) Co., Ltd., Shenzhen, China 5 Institute of Endemic Disease, Center for Disease Control and Prevention of Sichuan Province, Chengdu, Sichuan, China 6 The Center of Heart Development, College of Life Sciences, Hunan Norma University, Changsha, China #, These authors contributed equally to this work. *, Authors for correspondence. NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. medRxiv preprint doi: https://doi.org/10.1101/2021.03.12.21253491; this version posted March 15, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. -

2.04.132 Genetic Testing for Limb-Girdle Muscular Dystrophies
Medical Policy MP 2.04.132 Genetic Testing for Limb-Girdle Muscular Dystrophies BCBSA Ref. Policy: 2.04.132 Related Policies Last Review: 05/27/2021 2.04.86 Genetic Testing for Duchenne and Becker Effective Date: 05/27/2021 Muscular Dystrophy Section: Medicine 2.04.105 Genetic Testing for Facioscapulohumeral Muscular Dystrophy 2.04.570 Genetic Counseling DISCLAIMER/INSTRUCTIONS FOR USE Medical policy provides general guidance for applying Blue Cross of Idaho benefit plans (for purposes of medical policy, the terms “benefit plan” and “member contract” are used interchangeably). Coverage decisions must reference the member specific benefit plan document. The terms of the member specific benefit plan document may be different than the standard benefit plan upon which this medical policy is based. If there is a conflict between a member specific benefit plan and the Blue Cross of Idaho’s standard benefit plan, the member specific benefit plan supersedes this medical policy. Any person applying this medical policy must identify member eligibility, the member specific benefit plan, and any related policies or guidelines prior to applying this medical policy, including the existence of any state or federal guidance that may be specific to a line of business. Blue Cross of Idaho medical policies are designed for informational purposes only and are not an authorization, explanation of benefits or a contract. Receipt of benefits is subject to satisfaction of all terms and conditions of the member specific benefit plan coverage. Blue Cross of Idaho reserves the sole discretionary right to modify all its policies and guidelines at any time. -
FII Voter Ledger
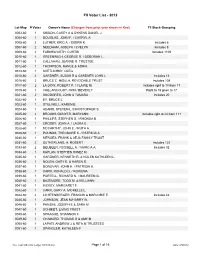
FII Voter List - 2012 Lot-Map # Votes Owner's Name (Changes from prior year shown in Red) FII Stock Grouping 0003-60 1 SISSON, CAREY A & DYKENS DANIEL J 0004-60 1 DOUGLAS, JOHN F. / CHERYL A. 0005-60 2 LUTHER, ERIC A. / DEBRA K. Includes 6 0007-60 2 NEEDHAM, JOSEPH / EVELYN Includes 8 0009-60 2 FARNSWORTH, CURTIS Includes 1109 0010-60 1 GREENWICH, GEORGE R. / DEBORAH L. 0011-60 1 CALLAHAN, JOANNE R. TRUSTEE 0012-60 1 THOMPSON, RANCE & ADRIA 0013-60 1 SVETLICHNY, OLEG 0015-60 2 GARDNER, SUSAN B & GARDNER JOHN L Includes 14 0016-60 2 BRUCE C. NISULA, REVOCABLE TRUST Includes 108 0017-60 2 LA DOW, ROBERT P. / CLAIRE M. Includes right to 18 from 17 0019-60 1 VAILLANCOURT, RON / BEVERLY Right to 18 given to 17 0021-60 2 SNODGRES, JOHN & TAMARA Includes 20 0022-60 1 EY, BRUCE L. 0023-60 1 STILLWELL, KAREN B. 0024-60 1 ADAMS, STEVEN L./CHRISTOPHER S. 0025-60 2 BROOKS-GONYER, MARYANN Includes right to 24 from 111 0026-60 1 PHILLIPS, STEPHEN S. / RHONDA B. 0027-60 1 CROSBY, JOHN A. / LAURA E. 0028-60 1 MCCARTHY, JOHN E. / RUTH A. 0029-60 1 POUNDS, THEODORE K. / PATRICIA A. 0030-60 1 MERGES, FRANK & AEJA FAMILY TRUST 0031-60 2 SUTHERLAND, H. ROBERT Includes 122 0033-60 2 DEUBLER, RUSSELL A. / MARCIA A. Includes 32 0034-60 1 KAPLAN, STEPHEN H/INEZ M 0035-60 1 GARDNER, KENNETH R. & NOLEN KATHLEEN L. 0036-60 1 NOLEN, GARY E. & KAREN R. 0037-60 1 DONOVAN, JOHN H. -

When 7 Transmembrane Receptors Are Not G Protein–Coupled Receptors
When 7 transmembrane receptors are not G protein–coupled receptors Keshava Rajagopal, … , Robert J. Lefkowitz, Howard A. Rockman J Clin Invest. 2005;115(11):2971-2974. https://doi.org/10.1172/JCI26950. Commentary Classically, 7 transmembrane receptors transduce extracellular signals by coupling to heterotrimeric G proteins, although recent in vitro studies have clearly demonstrated that they can also signal via G protein–independent mechanisms. However, the physiologic consequences of this unconventional signaling, particularly in vivo, have not been explored. In this issue of the JCI, Zhai et al. demonstrate in vivo effects of G protein–independent signaling by the angiotensin II type 1 receptor (AT1R). In studies of the mouse heart, they compare the physiologic and biochemical consequences of transgenic cardiac-specific overexpression of a mutant AT1R incapable of G protein coupling with those of a wild-type receptor. Their results not only provide the first glimpse of the physiologic effects of this newly appreciated mode of signaling but also provide important and previously unappreciated clues as to the underlying molecular mechanisms. Find the latest version: https://jci.me/26950/pdf commentaries gene, spastin, regulates microtubule stability to 16. Wittmann, C.W., et al. 2001. Tauopathy in Dro- 21. Chan, Y.B., et al. 2003. Neuromuscular defects in modulate synaptic structure and function. Curr. sophila: neurodegeneration without neurofibril- a Drosophila survival motor neuron gene mutant. Biol. 14:1135–1147. lary tangles. Science. 293:711–714. Hum. Mol. Genet. 12:1367–1376. 12. Sherwood, N.T., Sun, Q., Xue, M., Zhang, B., and 17. Shapiro, W.R., and Young, D.F.