Identifying Drug Sensitivity Subnetworks with NETPHIX
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Variation Across the Human Olfactory Receptor Repertoire Alters Odor Perception
bioRxiv preprint doi: https://doi.org/10.1101/212431; this version posted November 1, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Genetic variation across the human olfactory receptor repertoire alters odor perception Casey Trimmer1,*, Andreas Keller2, Nicolle R. Murphy1, Lindsey L. Snyder1, Jason R. Willer3, Maira Nagai4,5, Nicholas Katsanis3, Leslie B. Vosshall2,6,7, Hiroaki Matsunami4,8, and Joel D. Mainland1,9 1Monell Chemical Senses Center, Philadelphia, Pennsylvania, USA 2Laboratory of Neurogenetics and Behavior, The Rockefeller University, New York, New York, USA 3Center for Human Disease Modeling, Duke University Medical Center, Durham, North Carolina, USA 4Department of Molecular Genetics and Microbiology, Duke University Medical Center, Durham, North Carolina, USA 5Department of Biochemistry, University of Sao Paulo, Sao Paulo, Brazil 6Howard Hughes Medical Institute, New York, New York, USA 7Kavli Neural Systems Institute, New York, New York, USA 8Department of Neurobiology and Duke Institute for Brain Sciences, Duke University Medical Center, Durham, North Carolina, USA 9Department of Neuroscience, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania, USA *[email protected] ABSTRACT The human olfactory receptor repertoire is characterized by an abundance of genetic variation that affects receptor response, but the perceptual effects of this variation are unclear. To address this issue, we sequenced the OR repertoire in 332 individuals and examined the relationship between genetic variation and 276 olfactory phenotypes, including the perceived intensity and pleasantness of 68 odorants at two concentrations, detection thresholds of three odorants, and general olfactory acuity. -

Supplemental Data
1 Supplementary Figure 1. Immunohistochemical distribution of urothelial cells, renal tubular cells, and interstitial cells stained 2 by uroplakin III, kidney specific protein, and vimentin, respectively. Magnification, ×100 (inset x400). Representative 3 micrographs were obtained from normal papillary tissues of CaOx stone formers. 1 Supplementary Table 1. Top 100 upregulated genes in papillary tissue of both Randall’s Plaque and normal mucosa with calcium oxalate stone formers compared to those with control patients. Fold Agilent ID Gene Symbol Description change A_23_P128868 OR11H12 Homo sapiens olfactory receptor, family 11, subfamily H, member 12 (OR11H12), mRNA [NM_001013354] 26.613 Homo sapiens killer cell immunoglobulin-like receptor, two domains, short cytoplasmic tail, 2 (KIR2DS2), mRNA A_23_P130815 KIR2DS2 26.224 [NM_012312] A_24_P402855 PROL1 Homo sapiens proline rich, lacrimal 1 (PROL1), mRNA [NM_021225] 23.733 A_24_P917306 ZNF385D zinc finger protein 385D [Source:HGNC Symbol;Acc:26191] [ENST00000494108] 23.050 A_33_P3260667 OR2T34 Homo sapiens olfactory receptor, family 2, subfamily T, member 34 (OR2T34), mRNA [NM_001001821] 20.948 A_33_P3259440 GOLGA6A Homo sapiens golgin A6 family, member A (GOLGA6A), mRNA [NM_001038640] 20.628 A_33_P3417281 MUC4 Homo sapiens mucin 4, cell surface associated (MUC4), transcript variant 1, mRNA [NM_018406] 20.610 A_24_P239176 MUC4 Homo sapiens mucin 4, cell surface associated (MUC4), transcript variant 1, mRNA [NM_018406] 19.965 A_21_P0006968 SFTA1P Homo sapiens surfactant associated 1, pseudogene -

Copy Number Variation in Fetal Alcohol Spectrum Disorder
Biochemistry and Cell Biology Copy number variation in fetal alcohol spectrum disorder Journal: Biochemistry and Cell Biology Manuscript ID bcb-2017-0241.R1 Manuscript Type: Article Date Submitted by the Author: 09-Nov-2017 Complete List of Authors: Zarrei, Mehdi; The Centre for Applied Genomics Hicks, Geoffrey G.; University of Manitoba College of Medicine, Regenerative Medicine Reynolds, James N.; Queen's University School of Medicine, Biomedical and Molecular SciencesDraft Thiruvahindrapuram, Bhooma; The Centre for Applied Genomics Engchuan, Worrawat; Hospital for Sick Children SickKids Learning Institute Pind, Molly; University of Manitoba College of Medicine, Regenerative Medicine Lamoureux, Sylvia; The Centre for Applied Genomics Wei, John; The Centre for Applied Genomics Wang, Zhouzhi; The Centre for Applied Genomics Marshall, Christian R.; The Centre for Applied Genomics Wintle, Richard; The Centre for Applied Genomics Chudley, Albert; University of Manitoba Scherer, Stephen W.; The Centre for Applied Genomics Is the invited manuscript for consideration in a Special Fetal Alcohol Spectrum Disorder Issue? : Keyword: Fetal alcohol spectrum disorder, FASD, copy number variations, CNV https://mc06.manuscriptcentral.com/bcb-pubs Page 1 of 354 Biochemistry and Cell Biology 1 Copy number variation in fetal alcohol spectrum disorder 2 Mehdi Zarrei,a Geoffrey G. Hicks,b James N. Reynolds,c,d Bhooma Thiruvahindrapuram,a 3 Worrawat Engchuan,a Molly Pind,b Sylvia Lamoureux,a John Wei,a Zhouzhi Wang,a Christian R. 4 Marshall,a Richard F. Wintle,a Albert E. Chudleye,f and Stephen W. Scherer,a,g 5 aThe Centre for Applied Genomics and Program in Genetics and Genome Biology, The Hospital 6 for Sick Children, Toronto, Ontario, Canada 7 bRegenerative Medicine Program, University of Manitoba, Winnipeg, Canada 8 cCentre for Neuroscience Studies, Queen's University, Kingston, Ontario, Canada. -

Produktinformation
Produktinformation Diagnostik & molekulare Diagnostik Laborgeräte & Service Zellkultur & Verbrauchsmaterial Forschungsprodukte & Biochemikalien Weitere Information auf den folgenden Seiten! See the following pages for more information! Lieferung & Zahlungsart Lieferung: frei Haus Bestellung auf Rechnung SZABO-SCANDIC Lieferung: € 10,- HandelsgmbH & Co KG Erstbestellung Vorauskassa Quellenstraße 110, A-1100 Wien T. +43(0)1 489 3961-0 Zuschläge F. +43(0)1 489 3961-7 [email protected] • Mindermengenzuschlag www.szabo-scandic.com • Trockeneiszuschlag • Gefahrgutzuschlag linkedin.com/company/szaboscandic • Expressversand facebook.com/szaboscandic SANTA CRUZ BIOTECHNOLOGY, INC. OR2M5 siRNA (h): sc-88564 BACKGROUND STORAGE AND RESUSPENSION Olfactory receptors are G protein-coupled receptor proteins that localize to the Store lyophilized siRNA duplex at -20° C with desiccant. Stable for at least cilia of olfactory sensory neurons where they display affinity for and bind to one year from the date of shipment. Once resuspended, store at -20° C, a variety of odor molecules. The genes encoding olfactory receptors comprise avoid contact with RNAses and repeated freeze thaw cycles. the largest family in the human genome. The binding of olfactory receptor Resuspend lyophilized siRNA duplex in 330 µl of the RNAse-free water proteins to odor molecules triggers a signal transduction cascade that leads provided. Resuspension of the siRNA duplex in 330 µl of RNAse-free water to the production of cAMP via an olfactory-enriched adenylate cyclase. This makes a 10 µM solution in a 10 µM Tris-HCl, pH 8.0, 20 mM NaCl, 1 mM event ultimately leads to transmission of action potentials to the brain and EDTA buffered solution. the subsequent perception of smell. -

Microarray-Based Analysis of Genes, Transcription Factors, and Epigenetic Modifications in Lung Cancer Exposed to Nitric Oxide
CANCER GENOMICS & PROTEOMICS 17 : 401-415 (2020) doi:10.21873/cgp.20199 Microarray-based Analysis of Genes, Transcription Factors, and Epigenetic Modifications in Lung Cancer Exposed to Nitric Oxide ARNATCHAI MAIUTHED 1, ORNJIRA PRAKHONGCHEEP 2,3 and PITHI CHANVORACHOTE 2,3 1Department of Pharmacology, Faculty of Pharmacy, Mahidol University, Bangkok, Thailand; 2Cell-based Drug and Health Product Development Research Unit, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok, Thailand; 3Department of Pharmacology and Physiology, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok, Thailand Abstract. Background/Aim: Nitric oxide (NO) is recognized NO exposure of lung cancer cells resulted in a change in as an important biological mediator that exerts several transcription factors (TFs) and epigenetic modifications human physiological functions. As its nature is an aqueous (histone modification and miRNA). Interestingly, NO soluble gas that can diffuse through cells and tissues, NO treatment was shown to potentiate cancer stem cell-related can affect cell signaling, the phenotype of cancer and modify genes and transcription factors Oct4, Klf4, and Myc. surrounding cells. The variety of effects of NO on cancer cell Conclusion: Through this comprehensive approach, the biology has convinced researchers to determine the defined present study illustrated the scheme of how NO affects mechanisms of these effects and how to control this mediator molecular events in lung cancer cells. for a better understanding as well as for therapeutic gain. Materials and Methods: We used bioinformatics and Lung cancer is one of the leading causes of cancer-related pharmacological experiments to elucidate the potential deaths worldwide (1, 2). Since the aggressive phenotypes regulation and underlying mechanisms of NO in non-small and treatment failures have been frequently observed in lung a lung cancer cell model. -

Incorporating Genetic Networks Into Case-Control Association Studies with High-Dimensional DNA Methylation Data Kipoong Kim and Hokeun Sun*
Kim and Sun BMC Bioinformatics (2019) 20:510 https://doi.org/10.1186/s12859-019-3040-x METHODOLOGY ARTICLE Open Access Incorporating genetic networks into case-control association studies with high-dimensional DNA methylation data Kipoong Kim and Hokeun Sun* Abstract Background: In human genetic association studies with high-dimensional gene expression data, it has been well known that statistical selection methods utilizing prior biological network knowledge such as genetic pathways and signaling pathways can outperform other methods that ignore genetic network structures in terms of true positive selection. In recent epigenetic research on case-control association studies, relatively many statistical methods have been proposed to identify cancer-related CpG sites and their corresponding genes from high-dimensional DNA methylation array data. However, most of existing methods are not designed to utilize genetic network information although methylation levels between linked genes in the genetic networks tend to be highly correlated with each other. Results: We propose new approach that combines data dimension reduction techniques with network-based regularization to identify outcome-related genes for analysis of high-dimensional DNA methylation data. In simulation studies, we demonstrated that the proposed approach overwhelms other statistical methods that do not utilize genetic network information in terms of true positive selection. We also applied it to the 450K DNA methylation array data of the four breast invasive carcinoma cancer subtypes from The Cancer Genome Atlas (TCGA) project. Conclusions: The proposed variable selection approach can utilize prior biological network information for analysis of high-dimensional DNA methylation array data. It first captures gene level signals from multiple CpG sites using data a dimension reduction technique and then performs network-based regularization based on biological network graph information. -

Anti-OR10V1 (Aa 280-309) Polyclonal Antibody (DPABH-21566) This Product Is for Research Use Only and Is Not Intended for Diagnostic Use
Anti-OR10V1 (aa 280-309) polyclonal antibody (DPABH-21566) This product is for research use only and is not intended for diagnostic use. PRODUCT INFORMATION Antigen Description Olfactory receptors interact with odorant molecules in the nose, to initiate a neuronal response that triggers the perception of a smell. The olfactory receptor proteins are members of a large family of G-protein-coupled receptors (GPCR) arising from single coding-exon genes. Olfactory receptors share a 7-transmembrane domain structure with many neurotransmitter and hormone receptors and are responsible for the recognition and G protein-mediated transduction of odorant signals. The olfactory receptor gene family is the largest in the genome. The nomenclature assigned to the olfactory receptor genes and proteins for this organism is independent of other organisms. Immunogen Synthetic peptide conjugated to KLH, corresponding to a region within C terminal amino acids 280-309 of Human OR10V1 (NP_001005324.1). Isotype IgG Source/Host Rabbit Species Reactivity Human Purification Immunogen affinity purified Conjugate Unconjugated Applications WB Format Liquid Size 400 μl Buffer Constituent: 99% PBS Preservative 0.09% Sodium Azide Storage Store at 4°C (up to 6 months). For long term storage store at -20°C GENE INFORMATION 45-1 Ramsey Road, Shirley, NY 11967, USA Email: [email protected] Tel: 1-631-624-4882 Fax: 1-631-938-8221 1 © Creative Diagnostics All Rights Reserved Gene Name OR10V1 olfactory receptor, family 10, subfamily V, member 2 [ Homo sapiens ] Official Symbol OR10V1 Synonyms OR10V1; olfactory receptor, family 10, subfamily V, member 1; OR11-256; olfactory receptor 10V1; olfactory receptor OR11-256; Entrez Gene ID 390201 Protein Refseq NP_001005324.1 UniProt ID Q8NGI7 Pathway GPCR downstream signaling; Olfactory transduction; Signal Transduction. -

Olfactory Receptors in Non-Chemosensory Tissues
BMB Reports Invited Mini Review Olfactory receptors in non-chemosensory tissues NaNa Kang & JaeHyung Koo* Department of Brain Science, Daegu Gyeongbuk Institute of Science and Technology (DGIST), Daegu 711-873, Korea Olfactory receptors (ORs) detect volatile chemicals that lead to freezing behavior (3-5). the initial perception of smell in the brain. The olfactory re- ORs are localized in the cilia of olfactory sensory neurons ceptor (OR) is the first protein that recognizes odorants in the (OSNs) in the olfactory epithelium (OE) and are activated by olfactory signal pathway and it is present in over 1,000 genes chemical cues, typically odorants at the molecular level, in mice. It is also the largest member of the G protein-coupled which lead to the perception of smell in the brain (6). receptors (GPCRs). Most ORs are extensively expressed in the Tremendous research was conducted since Buck and Axel iso- nasal olfactory epithelium where they perform the appropriate lated ORs as an OE-specific expression in 1991 (7). OR genes, physiological functions that fit their location. However, recent the largest family among the G protein-coupled receptors whole-genome sequencing shows that ORs have been found (GPCRs) (8), constitute more than 1,000 genes on the mouse outside of the olfactory system, suggesting that ORs may play chromosome (9, 10) and more than 450 genes in the human an important role in the ectopic expression of non-chemo- genome (11, 12). sensory tissues. The ectopic expressions of ORs and their phys- Odorant activation shows a distinct signal transduction iological functions have attracted more attention recently since pathway for odorant perception. -

Sean Raspet – Molecules
1. Commercial name: Fructaplex© IUPAC Name: 2-(3,3-dimethylcyclohexyl)-2,5,5-trimethyl-1,3-dioxane SMILES: CC1(C)CCCC(C1)C2(C)OCC(C)(C)CO2 Molecular weight: 240.39 g/mol Volume (cubic Angstroems): 258.88 Atoms number (non-hydrogen): 17 miLogP: 4.43 Structure: Biological Properties: Predicted Druglikenessi: GPCR ligand -0.23 Ion channel modulator -0.03 Kinase inhibitor -0.6 Nuclear receptor ligand 0.15 Protease inhibitor -0.28 Enzyme inhibitor 0.15 Commercial name: Fructaplex© IUPAC Name: 2-(3,3-dimethylcyclohexyl)-2,5,5-trimethyl-1,3-dioxane SMILES: CC1(C)CCCC(C1)C2(C)OCC(C)(C)CO2 Predicted Olfactory Receptor Activityii: OR2L13 83.715% OR1G1 82.761% OR10J5 80.569% OR2W1 78.180% OR7A2 77.696% 2. Commercial name: Sylvoxime© IUPAC Name: N-[4-(1-ethoxyethenyl)-3,3,5,5tetramethylcyclohexylidene]hydroxylamine SMILES: CCOC(=C)C1C(C)(C)CC(CC1(C)C)=NO Molecular weight: 239.36 Volume (cubic Angstroems): 252.83 Atoms number (non-hydrogen): 17 miLogP: 4.33 Structure: Biological Properties: Predicted Druglikeness: GPCR ligand -0.6 Ion channel modulator -0.41 Kinase inhibitor -0.93 Nuclear receptor ligand -0.17 Protease inhibitor -0.39 Enzyme inhibitor 0.01 Commercial name: Sylvoxime© IUPAC Name: N-[4-(1-ethoxyethenyl)-3,3,5,5tetramethylcyclohexylidene]hydroxylamine SMILES: CCOC(=C)C1C(C)(C)CC(CC1(C)C)=NO Predicted Olfactory Receptor Activity: OR52D1 71.900% OR1G1 70.394% 0R52I2 70.392% OR52I1 70.390% OR2Y1 70.378% 3. Commercial name: Hyperflor© IUPAC Name: 2-benzyl-1,3-dioxan-5-one SMILES: O=C1COC(CC2=CC=CC=C2)OC1 Molecular weight: 192.21 g/mol Volume -
Supplementary Table S1. Genes Printed in the HC5 Microarray Employed in the Screening Phase of the Present Work
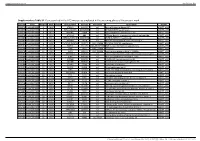
Supplementary material Ann Rheum Dis Supplementary Table S1. Genes printed in the HC5 microarray employed in the screening phase of the present work. CloneID Plate Position Well Length GeneSymbol GeneID Accession Description Vector 692672 HsxXG013989 2 B01 STK32A 202374 null serine/threonine kinase 32A pANT7_cGST 692675 HsxXG013989 3 C01 RPS10-NUDT3 100529239 null RPS10-NUDT3 readthrough pANT7_cGST 692678 HsxXG013989 4 D01 SPATA6L 55064 null spermatogenesis associated 6-like pANT7_cGST 692679 HsxXG013989 5 E01 ATP1A4 480 null ATPase, Na+/K+ transporting, alpha 4 polypeptide pANT7_cGST 692689 HsxXG013989 6 F01 ZNF816-ZNF321P 100529240 null ZNF816-ZNF321P readthrough pANT7_cGST 692691 HsxXG013989 7 G01 NKAIN1 79570 null Na+/K+ transporting ATPase interacting 1 pANT7_cGST 693155 HsxXG013989 8 H01 TNFSF12-TNFSF13 407977 NM_172089 TNFSF12-TNFSF13 readthrough pANT7_cGST 693161 HsxXG013989 9 A02 RAB12 201475 NM_001025300 RAB12, member RAS oncogene family pANT7_cGST 693169 HsxXG013989 10 B02 SYN1 6853 NM_133499 synapsin I pANT7_cGST 693176 HsxXG013989 11 C02 GJD3 125111 NM_152219 gap junction protein, delta 3, 31.9kDa pANT7_cGST 693181 HsxXG013989 12 D02 CHCHD10 400916 null coiled-coil-helix-coiled-coil-helix domain containing 10 pANT7_cGST 693184 HsxXG013989 13 E02 IDNK 414328 null idnK, gluconokinase homolog (E. coli) pANT7_cGST 693187 HsxXG013989 14 F02 LYPD6B 130576 null LY6/PLAUR domain containing 6B pANT7_cGST 693189 HsxXG013989 15 G02 C8orf86 389649 null chromosome 8 open reading frame 86 pANT7_cGST 693194 HsxXG013989 16 H02 CENPQ 55166 -
Quantitative-PCR Validation of 154 Genomic Segments Called As Cnvs in Five Replicat
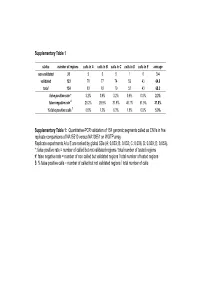
Supplementary Table 1 status number of regions calls in A calls in B calls in C calls in D calls in E average non validated 31 5 6 5 1 0 3.4 validated 123 78 77 74 52 43 64.8 total 154 83 83 79 53 43 68.2 false positive rate * 3.2% 3.9% 3.2% 0.6% 0.0% 2.2% false negative rate # 29.2% 29.9% 31.8% 46.1% 51.9% 37.8% % false positive calls $ 6.0% 7.2% 6.3% 1.9% 0.0% 5.0% Supplementary Table 1: Quantitative-PCR validation of 154 genomic segments called as CNVs in five replicate comparisons of NA15510 versus NA10851 on WGTP array Replicate experiments A to E are ranked by global SDe (A: 0.033; B: 0.033; C: 0.036; D: 0.039; E: 0.053). *: false positive rate = number of called but not validated regions / total number of tested regions #: false negative rate = number of non called but validated regions / total number of tested regions $: % false positive calls = number of called but not validated regions / total number of calls False positive estimates for 500K EA CNV calls Total Rep1 Rep2 Rep3 Avg (unique) Validated 33 28 32 31 38 Not validated 2 2 2 2 5 Total 35 30 34 33 43 % False positive 5.71% 6.67% 5.88% 6.09% - % False negative 13.16% 26.32% 15.79% 18.42% - Supplementary Table 2A : Quantitative PCR validation of 43 unique CNV regions called as CNVs in three replicate comparisons of NA15510 versus NA10851 using the 500K EA array. -

Table S3. RAE Analysis of Well-Differentiated Liposarcoma
Table S3. RAE analysis of well-differentiated liposarcoma Model Chromosome Region start Region end Size q value freqX0* # genes Genes Amp 1 145009467 145122002 112536 0.097 21.8 2 PRKAB2,PDIA3P Amp 1 145224467 146188434 963968 0.029 23.6 10 CHD1L,BCL9,ACP6,GJA5,GJA8,GPR89B,GPR89C,PDZK1P1,RP11-94I2.2,NBPF11 Amp 1 147475854 148412469 936616 0.034 23.6 20 PPIAL4A,FCGR1A,HIST2H2BF,HIST2H3D,HIST2H2AA4,HIST2H2AA3,HIST2H3A,HIST2H3C,HIST2H4B,HIST2H4A,HIST2H2BE, HIST2H2AC,HIST2H2AB,BOLA1,SV2A,SF3B4,MTMR11,OTUD7B,VPS45,PLEKHO1 Amp 1 148582896 153398462 4815567 1.5E-05 49.1 152 PRPF3,RPRD2,TARS2,ECM1,ADAMTSL4,MCL1,ENSA,GOLPH3L,HORMAD1,CTSS,CTSK,ARNT,SETDB1,LASS2,ANXA9, FAM63A,PRUNE,BNIPL,C1orf56,CDC42SE1,MLLT11,GABPB2,SEMA6C,TNFAIP8L2,LYSMD1,SCNM1,TMOD4,VPS72, PIP5K1A,PSMD4,ZNF687,PI4KB,RFX5,SELENBP1,PSMB4,POGZ,CGN,TUFT1,SNX27,TNRC4,MRPL9,OAZ3,TDRKH,LINGO4, RORC,THEM5,THEM4,S100A10,S100A11,TCHHL1,TCHH,RPTN,HRNR,FLG,FLG2,CRNN,LCE5A,CRCT1,LCE3E,LCE3D,LCE3C,LCE3B, LCE3A,LCE2D,LCE2C,LCE2B,LCE2A,LCE4A,KPRP,LCE1F,LCE1E,LCE1D,LCE1C,LCE1B,LCE1A,SMCP,IVL,SPRR4,SPRR1A,SPRR3, SPRR1B,SPRR2D,SPRR2A,SPRR2B,SPRR2E,SPRR2F,SPRR2C,SPRR2G,LELP1,LOR,PGLYRP3,PGLYRP4,S100A9,S100A12,S100A8, S100A7A,S100A7L2,S100A7,S100A6,S100A5,S100A4,S100A3,S100A2,S100A16,S100A14,S100A13,S100A1,C1orf77,SNAPIN,ILF2, NPR1,INTS3,SLC27A3,GATAD2B,DENND4B,CRTC2,SLC39A1,CREB3L4,JTB,RAB13,RPS27,NUP210L,TPM3,C1orf189,C1orf43,UBAP2L,HAX1, AQP10,ATP8B2,IL6R,SHE,TDRD10,UBE2Q1,CHRNB2,ADAR,KCNN3,PMVK,PBXIP1,PYGO2,SHC1,CKS1B,FLAD1,LENEP,ZBTB7B,DCST2, DCST1,ADAM15,EFNA4,EFNA3,EFNA1,RAG1AP1,DPM3 Amp 1