A Framework to Identify Contributing Genes In
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Variation Across the Human Olfactory Receptor Repertoire Alters Odor Perception
bioRxiv preprint doi: https://doi.org/10.1101/212431; this version posted November 1, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Genetic variation across the human olfactory receptor repertoire alters odor perception Casey Trimmer1,*, Andreas Keller2, Nicolle R. Murphy1, Lindsey L. Snyder1, Jason R. Willer3, Maira Nagai4,5, Nicholas Katsanis3, Leslie B. Vosshall2,6,7, Hiroaki Matsunami4,8, and Joel D. Mainland1,9 1Monell Chemical Senses Center, Philadelphia, Pennsylvania, USA 2Laboratory of Neurogenetics and Behavior, The Rockefeller University, New York, New York, USA 3Center for Human Disease Modeling, Duke University Medical Center, Durham, North Carolina, USA 4Department of Molecular Genetics and Microbiology, Duke University Medical Center, Durham, North Carolina, USA 5Department of Biochemistry, University of Sao Paulo, Sao Paulo, Brazil 6Howard Hughes Medical Institute, New York, New York, USA 7Kavli Neural Systems Institute, New York, New York, USA 8Department of Neurobiology and Duke Institute for Brain Sciences, Duke University Medical Center, Durham, North Carolina, USA 9Department of Neuroscience, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania, USA *[email protected] ABSTRACT The human olfactory receptor repertoire is characterized by an abundance of genetic variation that affects receptor response, but the perceptual effects of this variation are unclear. To address this issue, we sequenced the OR repertoire in 332 individuals and examined the relationship between genetic variation and 276 olfactory phenotypes, including the perceived intensity and pleasantness of 68 odorants at two concentrations, detection thresholds of three odorants, and general olfactory acuity. -

Copy Number Variation in Fetal Alcohol Spectrum Disorder
Biochemistry and Cell Biology Copy number variation in fetal alcohol spectrum disorder Journal: Biochemistry and Cell Biology Manuscript ID bcb-2017-0241.R1 Manuscript Type: Article Date Submitted by the Author: 09-Nov-2017 Complete List of Authors: Zarrei, Mehdi; The Centre for Applied Genomics Hicks, Geoffrey G.; University of Manitoba College of Medicine, Regenerative Medicine Reynolds, James N.; Queen's University School of Medicine, Biomedical and Molecular SciencesDraft Thiruvahindrapuram, Bhooma; The Centre for Applied Genomics Engchuan, Worrawat; Hospital for Sick Children SickKids Learning Institute Pind, Molly; University of Manitoba College of Medicine, Regenerative Medicine Lamoureux, Sylvia; The Centre for Applied Genomics Wei, John; The Centre for Applied Genomics Wang, Zhouzhi; The Centre for Applied Genomics Marshall, Christian R.; The Centre for Applied Genomics Wintle, Richard; The Centre for Applied Genomics Chudley, Albert; University of Manitoba Scherer, Stephen W.; The Centre for Applied Genomics Is the invited manuscript for consideration in a Special Fetal Alcohol Spectrum Disorder Issue? : Keyword: Fetal alcohol spectrum disorder, FASD, copy number variations, CNV https://mc06.manuscriptcentral.com/bcb-pubs Page 1 of 354 Biochemistry and Cell Biology 1 Copy number variation in fetal alcohol spectrum disorder 2 Mehdi Zarrei,a Geoffrey G. Hicks,b James N. Reynolds,c,d Bhooma Thiruvahindrapuram,a 3 Worrawat Engchuan,a Molly Pind,b Sylvia Lamoureux,a John Wei,a Zhouzhi Wang,a Christian R. 4 Marshall,a Richard F. Wintle,a Albert E. Chudleye,f and Stephen W. Scherer,a,g 5 aThe Centre for Applied Genomics and Program in Genetics and Genome Biology, The Hospital 6 for Sick Children, Toronto, Ontario, Canada 7 bRegenerative Medicine Program, University of Manitoba, Winnipeg, Canada 8 cCentre for Neuroscience Studies, Queen's University, Kingston, Ontario, Canada. -

Misexpression of Cancer/Testis (Ct) Genes in Tumor Cells and the Potential Role of Dream Complex and the Retinoblastoma Protein Rb in Soma-To-Germline Transformation
Michigan Technological University Digital Commons @ Michigan Tech Dissertations, Master's Theses and Master's Reports 2019 MISEXPRESSION OF CANCER/TESTIS (CT) GENES IN TUMOR CELLS AND THE POTENTIAL ROLE OF DREAM COMPLEX AND THE RETINOBLASTOMA PROTEIN RB IN SOMA-TO-GERMLINE TRANSFORMATION SABHA M. ALHEWAT Michigan Technological University, [email protected] Copyright 2019 SABHA M. ALHEWAT Recommended Citation ALHEWAT, SABHA M., "MISEXPRESSION OF CANCER/TESTIS (CT) GENES IN TUMOR CELLS AND THE POTENTIAL ROLE OF DREAM COMPLEX AND THE RETINOBLASTOMA PROTEIN RB IN SOMA-TO- GERMLINE TRANSFORMATION", Open Access Master's Thesis, Michigan Technological University, 2019. https://doi.org/10.37099/mtu.dc.etdr/933 Follow this and additional works at: https://digitalcommons.mtu.edu/etdr Part of the Cancer Biology Commons, and the Cell Biology Commons MISEXPRESSION OF CANCER/TESTIS (CT) GENES IN TUMOR CELLS AND THE POTENTIAL ROLE OF DREAM COMPLEX AND THE RETINOBLASTOMA PROTEIN RB IN SOMA-TO-GERMLINE TRANSFORMATION By Sabha Salem Alhewati A THESIS Submitted in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE In Biological Sciences MICHIGAN TECHNOLOGICAL UNIVERSITY 2019 © 2019 Sabha Alhewati This thesis has been approved in partial fulfillment of the requirements for the Degree of MASTER OF SCIENCE in Biological Sciences. Department of Biological Sciences Thesis Advisor: Paul Goetsch. Committee Member: Ebenezer Tumban. Committee Member: Zhiying Shan. Department Chair: Chandrashekhar Joshi. Table of Contents List of figures .......................................................................................................................v -

Predicting Human Olfactory Perception from Activities of Odorant Receptors
iScience ll OPEN ACCESS Article Predicting Human Olfactory Perception from Activities of Odorant Receptors Joel Kowalewski, Anandasankar Ray [email protected] odor perception HIGHLIGHTS Machine learning predicted activity of 34 human ORs for ~0.5 million chemicals chemical structure Activities of human ORs predicts OR activity could predict odor character using machine learning Few OR activities were needed to optimize r predictions of each odor e t c percept a AI r a odorant activates mul- h Behavior predictions in c Drosophila also need few r tiple ORs o olfactory receptor d o activities ts ic ed pr ity tiv ac OR Kowalewski & Ray, iScience 23, 101361 August 21, 2020 ª 2020 The Author(s). https://doi.org/10.1016/ j.isci.2020.101361 iScience ll OPEN ACCESS Article Predicting Human Olfactory Perception from Activities of Odorant Receptors Joel Kowalewski1 and Anandasankar Ray1,2,3,* SUMMARY Odor perception in humans is initiated by activation of odorant receptors (ORs) in the nose. However, the ORs linked to specific olfactory percepts are unknown, unlike in vision or taste where receptors are linked to perception of different colors and tastes. The large family of ORs (~400) and multiple receptors activated by an odorant present serious challenges. Here, we first use machine learning to screen ~0.5 million compounds for new ligands and identify enriched structural motifs for ligands of 34 human ORs. We next demonstrate that the activity of ORs successfully predicts many of the 146 different perceptual qualities of chem- icals. Although chemical features have been used to model odor percepts, we show that biologically relevant OR activity is often superior. -

Sean Raspet – Molecules
1. Commercial name: Fructaplex© IUPAC Name: 2-(3,3-dimethylcyclohexyl)-2,5,5-trimethyl-1,3-dioxane SMILES: CC1(C)CCCC(C1)C2(C)OCC(C)(C)CO2 Molecular weight: 240.39 g/mol Volume (cubic Angstroems): 258.88 Atoms number (non-hydrogen): 17 miLogP: 4.43 Structure: Biological Properties: Predicted Druglikenessi: GPCR ligand -0.23 Ion channel modulator -0.03 Kinase inhibitor -0.6 Nuclear receptor ligand 0.15 Protease inhibitor -0.28 Enzyme inhibitor 0.15 Commercial name: Fructaplex© IUPAC Name: 2-(3,3-dimethylcyclohexyl)-2,5,5-trimethyl-1,3-dioxane SMILES: CC1(C)CCCC(C1)C2(C)OCC(C)(C)CO2 Predicted Olfactory Receptor Activityii: OR2L13 83.715% OR1G1 82.761% OR10J5 80.569% OR2W1 78.180% OR7A2 77.696% 2. Commercial name: Sylvoxime© IUPAC Name: N-[4-(1-ethoxyethenyl)-3,3,5,5tetramethylcyclohexylidene]hydroxylamine SMILES: CCOC(=C)C1C(C)(C)CC(CC1(C)C)=NO Molecular weight: 239.36 Volume (cubic Angstroems): 252.83 Atoms number (non-hydrogen): 17 miLogP: 4.33 Structure: Biological Properties: Predicted Druglikeness: GPCR ligand -0.6 Ion channel modulator -0.41 Kinase inhibitor -0.93 Nuclear receptor ligand -0.17 Protease inhibitor -0.39 Enzyme inhibitor 0.01 Commercial name: Sylvoxime© IUPAC Name: N-[4-(1-ethoxyethenyl)-3,3,5,5tetramethylcyclohexylidene]hydroxylamine SMILES: CCOC(=C)C1C(C)(C)CC(CC1(C)C)=NO Predicted Olfactory Receptor Activity: OR52D1 71.900% OR1G1 70.394% 0R52I2 70.392% OR52I1 70.390% OR2Y1 70.378% 3. Commercial name: Hyperflor© IUPAC Name: 2-benzyl-1,3-dioxan-5-one SMILES: O=C1COC(CC2=CC=CC=C2)OC1 Molecular weight: 192.21 g/mol Volume -
Quantitative-PCR Validation of 154 Genomic Segments Called As Cnvs in Five Replicat
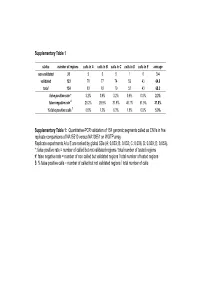
Supplementary Table 1 status number of regions calls in A calls in B calls in C calls in D calls in E average non validated 31 5 6 5 1 0 3.4 validated 123 78 77 74 52 43 64.8 total 154 83 83 79 53 43 68.2 false positive rate * 3.2% 3.9% 3.2% 0.6% 0.0% 2.2% false negative rate # 29.2% 29.9% 31.8% 46.1% 51.9% 37.8% % false positive calls $ 6.0% 7.2% 6.3% 1.9% 0.0% 5.0% Supplementary Table 1: Quantitative-PCR validation of 154 genomic segments called as CNVs in five replicate comparisons of NA15510 versus NA10851 on WGTP array Replicate experiments A to E are ranked by global SDe (A: 0.033; B: 0.033; C: 0.036; D: 0.039; E: 0.053). *: false positive rate = number of called but not validated regions / total number of tested regions #: false negative rate = number of non called but validated regions / total number of tested regions $: % false positive calls = number of called but not validated regions / total number of calls False positive estimates for 500K EA CNV calls Total Rep1 Rep2 Rep3 Avg (unique) Validated 33 28 32 31 38 Not validated 2 2 2 2 5 Total 35 30 34 33 43 % False positive 5.71% 6.67% 5.88% 6.09% - % False negative 13.16% 26.32% 15.79% 18.42% - Supplementary Table 2A : Quantitative PCR validation of 43 unique CNV regions called as CNVs in three replicate comparisons of NA15510 versus NA10851 using the 500K EA array. -

Supplemental Files 1-5.Pdf
Supplemental file 1: The whole list of tumor aberrations by time tumor biopsy ctDNA 2002 (T1) 2012 (M1) 2015 (M2) 08/2015 09/2015 11/2015 02/2016 05/2016 07/2016 11/2016 02/2017 05/2017 07/2017 11/2017 PIK3CA Q546R PIK3CA Q546R PIK3CA Q546R PIK3CA Q546R 0.30% PIK3CA Q546R 1.30% PIK3CA Q546R 5.40% PIK3CA Q546R 12.50% PIK3CA Q546R 9.50% PIK3CA Q546R 32.58% PIK3CA Q546R 44.30% PIK3CA Q546R 15.40% PIK3CA Q546R 41.00% TP53 E180K TP53 E180K TP53 E180K TP53 E180K 0.30% TP53 E180K 1.00% TP53 E180K 4.80% PIK3CA M1043I 0.20% PIK3CA M1043I 0.70% PIK3CA E453K 0.69% PIK3CA E453K 3.20% PIK3CA E453K 0.55% PIK3CA M1043I 0.20% PIK3R2 S688* PIK3R2 S688* ATR E2579K VHL E173Q 0.40% PTEN Q245* 0.20% PTEN Q245* 0.30% PIK3CA E726K 1.50% PIK3CA E245Q 0.10% PIK3CA M1043I 0.36% PIK3CA M1043I 1.20% TP53 E180K 11.80% PIK3CA E453K 0.25% PLCG2 P737T PLCG2 P737T PIK3CA E726K VHL E173Q 1.10% RB1 R556* 3.10% TP53 E180K 11.00% PIK3CA E726K 0.20% PIK3CA E726K 0.23% PIK3CA E726K 0.44% TP53 I195F 1.20% PIK3CA E39Q 0.53% CDKN1B Q163* PIK3CA M1004I MET S1061F MET L229F 0.30% CCND1 E9K 0.10% PTEN Q245* 0.80% TP53 E180K 7.30% TP53 E180K 24.28% TP53 E180K 35.40% TP53 L137Q 1.30% TP53 E180K 38.40% ERBB2 R487W PTEN S229* TBX3 S435* MET W112C 1.20% NF1 D1556N 1.30% TP53 I195F 0.20% TP53 R175G 0.20% TP53 I195F 1.70% ESR1 E380Q 7.10% TP53 I195F 0.56% ALK E1407K RB1 Q217* SPEN Q743* NF1 R1176T 1.00% NF1 E76K 0.30% ESR1 E380Q 0.90% TP53 I195F 1.37% TP53 L137Q 2.10% ESR1 L391V 0.41% TP53 I254M 0.25% APC E2637K PIK3CA D1017N PTEN I253M VHL E173Q 1.20% RB1 R556* 7.20% NF1 R1176T 0.50% TP53 -

Clinical, Molecular, and Immune Analysis of Dabrafenib-Trametinib
Supplementary Online Content Chen G, McQuade JL, Panka DJ, et al. Clinical, molecular and immune analysis of dabrafenib-trametinib combination treatment for metastatic melanoma that progressed during BRAF inhibitor monotherapy: a phase 2 clinical trial. JAMA Oncology. Published online April 28, 2016. doi:10.1001/jamaoncol.2016.0509. eMethods. eReferences. eTable 1. Clinical efficacy eTable 2. Adverse events eTable 3. Correlation of baseline patient characteristics with treatment outcomes eTable 4. Patient responses and baseline IHC results eFigure 1. Kaplan-Meier analysis of overall survival eFigure 2. Correlation between IHC and RNAseq results eFigure 3. pPRAS40 expression and PFS eFigure 4. Baseline and treatment-induced changes in immune infiltrates eFigure 5. PD-L1 expression eTable 5. Nonsynonymous mutations detected by WES in baseline tumors This supplementary material has been provided by the authors to give readers additional information about their work. © 2016 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 09/30/2021 eMethods Whole exome sequencing Whole exome capture libraries for both tumor and normal samples were constructed using 100ng genomic DNA input and following the protocol as described by Fisher et al.,3 with the following adapter modification: Illumina paired end adapters were replaced with palindromic forked adapters with unique 8 base index sequences embedded within the adapter. In-solution hybrid selection was performed using the Illumina Rapid Capture Exome enrichment kit with 38Mb target territory (29Mb baited). The targeted region includes 98.3% of the intervals in the Refseq exome database. Dual-indexed libraries were pooled into groups of up to 96 samples prior to hybridization. -

A Framework to Identify Contributing Genes in Patients with Phelan-Mcdermid Syndrome
www.nature.com/npjgenmed Corrected: Author Correction ARTICLE OPEN A framework to identify contributing genes in patients with Phelan-McDermid syndrome Anne-Claude Tabet 1,2,3,4, Thomas Rolland2,3,4, Marie Ducloy2,3,4, Jonathan Lévy1, Julien Buratti2,3,4, Alexandre Mathieu2,3,4, Damien Haye1, Laurence Perrin1, Céline Dupont 1, Sandrine Passemard1, Yline Capri1, Alain Verloes1, Séverine Drunat1, Boris Keren5, Cyril Mignot6, Isabelle Marey7, Aurélia Jacquette7, Sandra Whalen7, Eva Pipiras8, Brigitte Benzacken8, Sandra Chantot-Bastaraud9, Alexandra Afenjar10, Delphine Héron10, Cédric Le Caignec11, Claire Beneteau11, Olivier Pichon11, Bertrand Isidor11, Albert David11, Laila El Khattabi12, Stephan Kemeny13, Laetitia Gouas13, Philippe Vago13, Anne-Laure Mosca-Boidron14, Laurence Faivre15, Chantal Missirian16, Nicole Philip16, Damien Sanlaville17, Patrick Edery18, Véronique Satre19, Charles Coutton19, Françoise Devillard19, Klaus Dieterich20, Marie-Laure Vuillaume21, Caroline Rooryck21, Didier Lacombe21, Lucile Pinson22, Vincent Gatinois22, Jacques Puechberty22, Jean Chiesa23, James Lespinasse24, Christèle Dubourg25, Chloé Quelin25, Mélanie Fradin25, Hubert Journel26, Annick Toutain27, Dominique Martin28, Abdelamdjid Benmansour1, Claire S. Leblond2,3,4, Roberto Toro2,3,4, Frédérique Amsellem29, Richard Delorme2,3,4,29 and Thomas Bourgeron2,3,4 Phelan-McDermid syndrome (PMS) is characterized by a variety of clinical symptoms with heterogeneous degrees of severity, including intellectual disability (ID), absent or delayed speech, and autism spectrum disorders (ASD). It results from a deletion of the distal part of chromosome 22q13 that in most cases includes the SHANK3 gene. SHANK3 is considered a major gene for PMS, but the factors that modulate the severity of the syndrome remain largely unknown. In this study, we investigated 85 patients with different 22q13 rearrangements (78 deletions and 7 duplications). -

OR1C1 (NM 012353) Human Tagged ORF Clone – RC216791L4
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RC216791L4 OR1C1 (NM_012353) Human Tagged ORF Clone Product data: Product Type: Expression Plasmids Product Name: OR1C1 (NM_012353) Human Tagged ORF Clone Tag: mGFP Symbol: OR1C1 Synonyms: HSTPCR27; OR1-42; OR1.5.10; ORL211; TPCR27 Vector: pLenti-C-mGFP-P2A-Puro (PS100093) E. coli Selection: Chloramphenicol (34 ug/mL) Cell Selection: Puromycin ORF Nucleotide The ORF insert of this clone is exactly the same as(RC216791). Sequence: Restriction Sites: SgfI-MluI Cloning Scheme: ACCN: NM_012353 ORF Size: 942 bp This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 OR1C1 (NM_012353) Human Tagged ORF Clone – RC216791L4 OTI Disclaimer: The molecular sequence of this clone aligns with the gene accession number as a point of reference only. However, individual transcript sequences of the same gene can differ through naturally occurring variations (e.g. polymorphisms), each with its own valid existence. This clone is substantially in agreement with the reference, but a complete review of all prevailing variants is recommended prior to use. More info OTI Annotation: This clone was engineered to express the complete ORF with an expression tag. Expression varies depending on the nature of the gene. RefSeq: NM_012353.1, NP_036485.2 RefSeq Size: 1143 bp RefSeq ORF: 945 bp Locus ID: 26188 UniProt ID: Q15619, A0A126GV94 Protein Pathways: Olfactory transduction MW: 34.9 kDa Gene Summary: Olfactory receptors interact with odorant molecules in the nose, to initiate a neuronal response that triggers the perception of a smell. -

Output Results of CLIME (Clustering by Inferred Models of Evolution)
Output results of CLIME (CLustering by Inferred Models of Evolution) Dataset: Num of genes in input gene set: 4 Total number of genes: 20834 Prediction LLR threshold: 0 The CLIME PDF output two sections: 1) Overview of Evolutionarily Conserved Modules (ECMs) Top panel shows the predefined species tree. Bottom panel shows the partition of input genes into Evolutionary Conserved Modules (ECMs), ordered by ECM strength (shown at right), and separated by horizontal lines. Each row show one gene, where the phylogenetic profile indicates presence (blue) or absence (gray) of homologs in each species (column). Gene symbols are shown at left. Gray color indicates that the gene is a paralog to a higher scoring gene within the same ECM (based on BLASTP E < 1e-3). 2) Details of each ECM and its expansion ECM+ Top panel shows the inferred evolutionary history on the predefined species tree. Branch color shows the gain event (blue) and loss events (red color, with brighter color indicating higher confidence in loss). Branches before the gain or after a loss are shown in gray. Bottom panel shows the input genes that are within the ECM (blue/white rows) as well as all genes in the expanded ECM+ (green/gray rows). The ECM+ includes genes likely to have arisen under the inferred model of evolution relative to a background model, and scored using a log likelihood ratio (LLR). PG indicates "paralog group" and are labeled alphabetically (i.e., A, B). The first gene within each paralog group is shown in black color. All other genes sharing sequence similarity (BLAST E < 1e-3) are assigned to the same PG label and displayed in gray. -

The Hypothalamus As a Hub for SARS-Cov-2 Brain Infection and Pathogenesis
bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.139329; this version posted June 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. The hypothalamus as a hub for SARS-CoV-2 brain infection and pathogenesis Sreekala Nampoothiri1,2#, Florent Sauve1,2#, Gaëtan Ternier1,2ƒ, Daniela Fernandois1,2 ƒ, Caio Coelho1,2, Monica ImBernon1,2, Eleonora Deligia1,2, Romain PerBet1, Vincent Florent1,2,3, Marc Baroncini1,2, Florence Pasquier1,4, François Trottein5, Claude-Alain Maurage1,2, Virginie Mattot1,2‡, Paolo GiacoBini1,2‡, S. Rasika1,2‡*, Vincent Prevot1,2‡* 1 Univ. Lille, Inserm, CHU Lille, Lille Neuroscience & Cognition, DistAlz, UMR-S 1172, Lille, France 2 LaBoratorY of Development and PlasticitY of the Neuroendocrine Brain, FHU 1000 daYs for health, EGID, School of Medicine, Lille, France 3 Nutrition, Arras General Hospital, Arras, France 4 Centre mémoire ressources et recherche, CHU Lille, LiCEND, Lille, France 5 Univ. Lille, CNRS, INSERM, CHU Lille, Institut Pasteur de Lille, U1019 - UMR 8204 - CIIL - Center for Infection and ImmunitY of Lille (CIIL), Lille, France. # and ƒ These authors contriButed equallY to this work. ‡ These authors directed this work *Correspondence to: [email protected] and [email protected] Short title: Covid-19: the hypothalamic hypothesis 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.139329; this version posted June 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.