Incorporating Genetic Networks Into Case-Control Association Studies with High-Dimensional DNA Methylation Data Kipoong Kim and Hokeun Sun*
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1 Genome-Wide Comparisons of Variation in Linkage Disequilibrium
Downloaded from genome.cshlp.org on September 30, 2021 - Published by Cold Spring Harbor Laboratory Press Genome-wide comparisons of variation in linkage disequilibrium Yik Y. Teo1,*, Andrew E. Fry1, Kanishka Bhattacharya1, Kerrin S. Small1, Dominic P. Kwiatkowski1,2, Taane G. Clark1,2 1 Wellcome Trust Centre for Human Genetics, University of Oxford, United Kingdom 2 Wellcome Trust Sanger Institute, Hinxton, United Kingdom Running title: Genome-wide comparisons of LD Keywords: linkage disequilibrium, imputation, positive selection, meta-analysis, genome-wide association study * Corresponding author: Wellcome Trust Centre for Human Genetics, Roosevelt Drive, Oxford OX3 7BN, United Kingdom. Email: [email protected], phone: +44 1865 287712, fax: +44 1865 287 501. ABSTRACT Current genome-wide surveys of common diseases and complex traits fundamentally aim to detect indirect associations where the SNPs carrying the association signals are not biologically active but are in linkage disequilibrium (LD) with some unknown functional polymorphisms. Reproducing any novel discoveries from these genome-wide scans in independent studies is now a prerequisite for the putative findings to be accepted. Significant differences in patterns of LD between populations can affect the portability of phenotypic associations when the replication effort or meta-analyses are attempted in populations that are distinct from the original population which the genome-wide study is performed in. Here we introduce a novel method for genome-wide analyses of LD variations between populations that allow the identification of candidate regions with different patterns of LD. The evidence of LD variation provided by the introduced method correlated with the degree of differences in the frequencies of the most common haplotype across the populations. -

Prognostic Classifier Based on Genome-Wide DNA Methylation Profiling in Well-Differentiated Thyroid Tumors
CLINICAL RESEARCH ARTICLE Downloaded from https://academic.oup.com/jcem/article-abstract/102/11/4089/4082866 by Universidade Estadual Paulista J�lio de Mesquita Filho user on 06 May 2019 Prognostic Classifier Based on Genome-Wide DNA Methylation Profiling in Well-Differentiated Thyroid Tumors Mariana Bisarro dos Reis,1,2* Mateus Camargo Barros-Filho,1* F´abio Albuquerque Marchi,1 Caroline Moraes Beltrami,1 Hellen Kuasne,1 Clovis ´ Antonioˆ Lopes Pinto,3 Srikant Ambatipudi,4,5 Zdenko Herceg,4 Luiz Paulo Kowalski,6 and Silvia Regina Rogatto2,7 1International Research Center, CIPE, A.C. Camargo Cancer Center and National Institute of Science and Technology in Oncogenomics, S~ao Paulo 01509-010, SP, Brazil; 2Department of Urology, Faculty of Medicine, UNESP, S~ao Paulo State University, Botucatu 18618-970, SP, Brazil; 3Department of Pathology, A.C. Camargo Cancer Center, S~ao Paulo 01509-010, SP, Brazil; 4Epigenetics Group; International Agency for Research on Cancer (IARC), Lyon 69372, France; 5MRC Integrative Epidemiology Unit, University of Bristol, Bristol BS8 1TH, United Kingdom; 6Department of Head and Neck Surgery and Otorhinolaryngology, A.C. Camargo Cancer Center, S~ao Paulo 01509-010, SP, Brazil; and 7Department of Clinical Genetics, Vejle Hospital and Institute of Regional Health Research, University of Southern Denmark, Vejle, 7100, Denmark Context: Even though the majority of well-differentiated thyroid carcinoma (WDTC) is indolent, a number of cases display an aggressive behavior. Cumulative evidence suggests that the deregulation of DNA methylation has the potential to point out molecular markers associated with worse prognosis. Objective: To identify a prognostic epigenetic signature in thyroid cancer. -

Durbinharly.Pdf (9.770Mb)
GENOMICS OF SEASONAL HAIR SHEDDING AND ECOREGION-SPECIFIC GROWTH TO IDENTIFY ENVIRONMENTALLY-ADAPTED BEEF CATTLE _______________________________________ A Dissertation presented to the Faculty of the Graduate School at the University of Missouri-Columbia _______________________________________________________ In Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy _____________________________________________________ by HARLY JANE DURBIN Dr. Jared Decker, Dissertation Supervisor December 2020 APPROVAL PAGE The undersigned, appointed by the Dean of the Graduate School, have examined the dissertation entitled: GENOMICS OF SEASONAL HAIR SHEDDING AND ECO-REGION SPECIFIC GROWTH TO IDENTIFY ENVIRONMENTALLY-ADAPTED BEEF CATTLE Presented by Harly Jane Durbin, a candidate for the degree of Doctor of Philosophy, and hereby certify that in their opinion it is worthy of acceptance. _____________________________________ Dr. Jared E. Decker, Animal Sciences, UMC _____________________________________ Dr. Robert D. Schnabel, Animal Sciences, UMC _____________________________________ Dr. Jeremy F. Taylor, Animal Sciences, UMC _____________________________________ Dr. Elizabeth G. King, Biological Sciences, UMC _____________________________________ Dr. Stephen P. Miller, Angus Genetics, Inc. DEDICATION Momma – thank you for teaching me compassion and also how to approach life with a sense of humor. You do such an incredible amount for other people and I strive to be as selfless every day. Daddy – thank you for encouraging me to think independently and for teaching me how to stand up for myself. Thank you also for the years of FFA (also known sometimes as Father Feeds Animal) and getting up early to haul heifers to jackpot shows. Darby – thank you for holding me to a high standard. I often think there was some mix-up and that you were meant to be the older sister for how much you hold me up. -
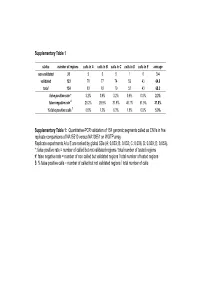
Quantitative-PCR Validation of 154 Genomic Segments Called As Cnvs in Five Replicat
Supplementary Table 1 status number of regions calls in A calls in B calls in C calls in D calls in E average non validated 31 5 6 5 1 0 3.4 validated 123 78 77 74 52 43 64.8 total 154 83 83 79 53 43 68.2 false positive rate * 3.2% 3.9% 3.2% 0.6% 0.0% 2.2% false negative rate # 29.2% 29.9% 31.8% 46.1% 51.9% 37.8% % false positive calls $ 6.0% 7.2% 6.3% 1.9% 0.0% 5.0% Supplementary Table 1: Quantitative-PCR validation of 154 genomic segments called as CNVs in five replicate comparisons of NA15510 versus NA10851 on WGTP array Replicate experiments A to E are ranked by global SDe (A: 0.033; B: 0.033; C: 0.036; D: 0.039; E: 0.053). *: false positive rate = number of called but not validated regions / total number of tested regions #: false negative rate = number of non called but validated regions / total number of tested regions $: % false positive calls = number of called but not validated regions / total number of calls False positive estimates for 500K EA CNV calls Total Rep1 Rep2 Rep3 Avg (unique) Validated 33 28 32 31 38 Not validated 2 2 2 2 5 Total 35 30 34 33 43 % False positive 5.71% 6.67% 5.88% 6.09% - % False negative 13.16% 26.32% 15.79% 18.42% - Supplementary Table 2A : Quantitative PCR validation of 43 unique CNV regions called as CNVs in three replicate comparisons of NA15510 versus NA10851 using the 500K EA array. -

Table S3. RAE Analysis of Well-Differentiated Liposarcoma
Table S3. RAE analysis of well-differentiated liposarcoma Model Chromosome Region start Region end Size q value freqX0* # genes Genes Amp 1 145009467 145122002 112536 0.097 21.8 2 PRKAB2,PDIA3P Amp 1 145224467 146188434 963968 0.029 23.6 10 CHD1L,BCL9,ACP6,GJA5,GJA8,GPR89B,GPR89C,PDZK1P1,RP11-94I2.2,NBPF11 Amp 1 147475854 148412469 936616 0.034 23.6 20 PPIAL4A,FCGR1A,HIST2H2BF,HIST2H3D,HIST2H2AA4,HIST2H2AA3,HIST2H3A,HIST2H3C,HIST2H4B,HIST2H4A,HIST2H2BE, HIST2H2AC,HIST2H2AB,BOLA1,SV2A,SF3B4,MTMR11,OTUD7B,VPS45,PLEKHO1 Amp 1 148582896 153398462 4815567 1.5E-05 49.1 152 PRPF3,RPRD2,TARS2,ECM1,ADAMTSL4,MCL1,ENSA,GOLPH3L,HORMAD1,CTSS,CTSK,ARNT,SETDB1,LASS2,ANXA9, FAM63A,PRUNE,BNIPL,C1orf56,CDC42SE1,MLLT11,GABPB2,SEMA6C,TNFAIP8L2,LYSMD1,SCNM1,TMOD4,VPS72, PIP5K1A,PSMD4,ZNF687,PI4KB,RFX5,SELENBP1,PSMB4,POGZ,CGN,TUFT1,SNX27,TNRC4,MRPL9,OAZ3,TDRKH,LINGO4, RORC,THEM5,THEM4,S100A10,S100A11,TCHHL1,TCHH,RPTN,HRNR,FLG,FLG2,CRNN,LCE5A,CRCT1,LCE3E,LCE3D,LCE3C,LCE3B, LCE3A,LCE2D,LCE2C,LCE2B,LCE2A,LCE4A,KPRP,LCE1F,LCE1E,LCE1D,LCE1C,LCE1B,LCE1A,SMCP,IVL,SPRR4,SPRR1A,SPRR3, SPRR1B,SPRR2D,SPRR2A,SPRR2B,SPRR2E,SPRR2F,SPRR2C,SPRR2G,LELP1,LOR,PGLYRP3,PGLYRP4,S100A9,S100A12,S100A8, S100A7A,S100A7L2,S100A7,S100A6,S100A5,S100A4,S100A3,S100A2,S100A16,S100A14,S100A13,S100A1,C1orf77,SNAPIN,ILF2, NPR1,INTS3,SLC27A3,GATAD2B,DENND4B,CRTC2,SLC39A1,CREB3L4,JTB,RAB13,RPS27,NUP210L,TPM3,C1orf189,C1orf43,UBAP2L,HAX1, AQP10,ATP8B2,IL6R,SHE,TDRD10,UBE2Q1,CHRNB2,ADAR,KCNN3,PMVK,PBXIP1,PYGO2,SHC1,CKS1B,FLAD1,LENEP,ZBTB7B,DCST2, DCST1,ADAM15,EFNA4,EFNA3,EFNA1,RAG1AP1,DPM3 Amp 1 -

Strong Selection During the Last Millennium for African Ancestry In
Strong selection during the last millennium for African ancestry in the admixed population of Madagascar Denis Pierron, Margit Heiske, Harilanto Razafindrazaka, Veronica Pereda-Loth, Jazmin Sanchez, Omar Alva, Amal Arachiche, Anne Boland, Robert Olaso, Jean-François Deleuze, et al. To cite this version: Denis Pierron, Margit Heiske, Harilanto Razafindrazaka, Veronica Pereda-Loth, Jazmin Sanchez, et al.. Strong selection during the last millennium for African ancestry in the admixed population of Mada- gascar. Nature Communications, Nature Publishing Group, 2018, 9 (1), pp.932. 10.1038/s41467-018- 03342-5. hal-02112693 HAL Id: hal-02112693 https://hal.archives-ouvertes.fr/hal-02112693 Submitted on 9 Jan 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License ARTICLE DOI: 10.1038/s41467-018-03342-5 OPEN Strong selection during the last millennium for African ancestry in the admixed population of Madagascar Denis Pierron1, Margit Heiske1, Harilanto Razafindrazaka2,1, Veronica Pereda-loth1, Jazmin Sanchez1, Omar Alva1, Amal Arachiche1, Anne Boland3, Robert Olaso3, Jean-Francois Deleuze3, Francois-Xavier Ricaut1, Jean-Aimé Rakotoarisoa4, Chantal Radimilahy4, Mark Stoneking5 & Thierry Letellier1 1234567890():,; While admixed populations offer a unique opportunity to detect selection, the admixture in most of the studied populations occurred too recently to produce conclusive signals. -
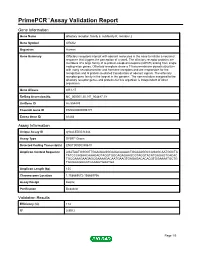
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name olfactory receptor, family 6, subfamily K, member 2 Gene Symbol OR6K2 Organism Human Gene Summary Olfactory receptors interact with odorant molecules in the nose to initiate a neuronal response that triggers the perception of a smell. The olfactory receptor proteins are members of a large family of G-protein-coupled receptors (GPCR) arising from single coding-exon genes. Olfactory receptors share a 7-transmembrane domain structure with many neurotransmitter and hormone receptors and are responsible for the recognition and G protein-mediated transduction of odorant signals. The olfactory receptor gene family is the largest in the genome. The nomenclature assigned to the olfactory receptor genes and proteins for this organism is independent of other organisms. Gene Aliases OR1-17 RefSeq Accession No. NC_000001.10, NT_004487.19 UniGene ID Hs.554489 Ensembl Gene ID ENSG00000196171 Entrez Gene ID 81448 Assay Information Unique Assay ID qHsaCED0034344 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000359610 Amplicon Context Sequence AGATAATGGGGTTGAAGAAGGGAGACAAAACTGCAAAGGCCAGAGCAATGGCTA TATCCCAGAACAAAGAGTAGGTGGCAGAGAAGCGTAGGTACATGAGAGTCACAC TGCCAAAGAAGAGCGAAAAGACAATGAAGTGAGAGACACACGTGGAAAATGCTG TGCGGCGGCCTCCAGCTGAATGA Amplicon Length (bp) 155 Chromosome Location 1:158669572-158669756 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 114 R2 0.9912 Page 1/5 PrimePCR™Assay Validation Report cDNA Cq 33.55 cDNA Tm (Celsius) 83 gDNA Cq 23.81 Specificity -

The Hypothalamus As a Hub for SARS-Cov-2 Brain Infection and Pathogenesis
bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.139329; this version posted June 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. The hypothalamus as a hub for SARS-CoV-2 brain infection and pathogenesis Sreekala Nampoothiri1,2#, Florent Sauve1,2#, Gaëtan Ternier1,2ƒ, Daniela Fernandois1,2 ƒ, Caio Coelho1,2, Monica ImBernon1,2, Eleonora Deligia1,2, Romain PerBet1, Vincent Florent1,2,3, Marc Baroncini1,2, Florence Pasquier1,4, François Trottein5, Claude-Alain Maurage1,2, Virginie Mattot1,2‡, Paolo GiacoBini1,2‡, S. Rasika1,2‡*, Vincent Prevot1,2‡* 1 Univ. Lille, Inserm, CHU Lille, Lille Neuroscience & Cognition, DistAlz, UMR-S 1172, Lille, France 2 LaBoratorY of Development and PlasticitY of the Neuroendocrine Brain, FHU 1000 daYs for health, EGID, School of Medicine, Lille, France 3 Nutrition, Arras General Hospital, Arras, France 4 Centre mémoire ressources et recherche, CHU Lille, LiCEND, Lille, France 5 Univ. Lille, CNRS, INSERM, CHU Lille, Institut Pasteur de Lille, U1019 - UMR 8204 - CIIL - Center for Infection and ImmunitY of Lille (CIIL), Lille, France. # and ƒ These authors contriButed equallY to this work. ‡ These authors directed this work *Correspondence to: [email protected] and [email protected] Short title: Covid-19: the hypothalamic hypothesis 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.139329; this version posted June 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Supplemental Table 1 (S1). the Chromosomal Regions and Genes Exhibiting Loss of Heterozygosity That Were Shared Between the HLRCC-Rccs of Both Patients 1 and 2
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Clin Pathol Supplemental Table 1 (S1). The chromosomal regions and genes exhibiting loss of heterozygosity that were shared between the HLRCC-RCCs of both Patients 1 and 2. Chromosome Position Genes 1 p13.1 ATP1A1, ATP1A1-AS1, LOC101929023, CD58, IGSF3, MIR320B1, C1orf137, CD2, PTGFRN, CD101, LOC101929099 1 p21.1* COL11A1, LOC101928436, RNPC3, AMY2B, ACTG1P4, AMY2A, AMY1A, AMY1C, AMY1B 1 p36.11 LOC101928728, ARID1A, PIGV, ZDHHC18, SFN, GPN2, GPATCH3, NR0B2, NUDC, KDF1, TRNP1, FAM46B, SLC9A1, WDTC1, TMEM222, ACTG1P20, SYTL1, MAP3K6, FCN3, CD164L2, GPR3, WASF2, AHDC1, FGR, IFI6 1 q21.3* KCNN3, PMVK, PBXIP1, PYGO2, LOC101928120, SHC1, CKS1B, MIR4258, FLAD1, LENEP, ZBTB7B, DCST2, DCST1, LOC100505666, ADAM15, EFNA4, EFNA3, EFNA1, SLC50A1, DPM3, KRTCAP2, TRIM46, MUC1, MIR92B, THBS3, MTX1, GBAP1, GBA, FAM189B, SCAMP3, CLK2, HCN3, PKLR, FDPS, RUSC1-AS1, RUSC1, ASH1L, MIR555, POU5F1P4, ASH1L-AS1, MSTO1, MSTO2P, YY1AP1, SCARNA26A, DAP3, GON4L, SCARNA26B, SYT11, RIT1, KIAA0907, SNORA80E, SCARNA4, RXFP4, ARHGEF2, MIR6738, SSR2, UBQLN4, LAMTOR2, RAB25, MEX3A, LMNA, SEMA4A, SLC25A44, PMF1, PMF1-BGLAP 1 q24.2–44* LOC101928650, GORAB, PRRX1, MROH9, FMO3, MIR1295A, MIR1295B, FMO6P, FMO2, FMO1, FMO4, TOP1P1, PRRC2C, MYOC, VAMP4, METTL13, DNM3, DNM3-IT1, DNM3OS, MIR214, MIR3120, MIR199A2, C1orf105, PIGC, SUCO, FASLG, TNFSF18, TNFSF4, LOC100506023, LOC101928673, -

Learning Natural Selection from the Site Frequency Spectrum a Letter Submitted to Genetics
Genetics: Early Online, published on June 21, 2013 as 10.1534/genetics.113.152587 Learning Natural Selection from the Site Frequency Spectrum A Letter Submitted to Genetics Roy Ronen∗, Nitin Udpa∗, Eran Halperiny z x, and Vineet Bafna{ June 15, 2013 ∗Bioinformatics & Systems Biology Program, University of California, San Diego, CA, USA yThe Blavatnik School of Computer Science, Tel-Aviv University, Tel-Aviv, Israel zInternational Computer Science Institute, Berkeley, CA, USA xDepartment of Molecular Microbiology & Biotechnology, Tel-Aviv University, Tel-Aviv, Israel {Department of Computer Science & Engineering, University of California, San Diego, CA, USA 1 Copyright 2013. Running Head: Learning Selection from the SFS Key Words: Natural Selection, Supervised Learning, Site Frequency Spectrum Corresponding Author: Roy Ronen Bioinformatics & Systems Biology Program University of California, San Diego 9500 Gilman Drive, Dept. 0419 La Jolla, CA 92093 (858) 534-8865 (ph.) (858) 534-7029 (fax.) [email protected] 2 Abstract Genetic adaptation to external stimuli occurs through the combined action of mutation and selection. A central problem in genetics is to identify loci responsive to specific selective constraints. Many tests have been proposed to identify the genomic signatures of natural selection by quantifying the skew in the site frequency spectrum (SFS) under selection rel- ative to neutrality. We build upon recent work that connects many of these tests under a common framework, by describing how selective sweeps impact the scaled SFS. We show that the specific skew depends on many attributes of the sweep, including the selection coef- ficient and the time under selection. Using supervised learning on extensive simulated data, we characterize the features of the scaled SFS that best separate different types of selective sweeps from neutrality. -

Supplementary Table 1
Supplementary Table 1. List of genes that encode proteins contianing cell surface epitopes and are represented on Agilent human 1A V2 microarray chip (2,177 genes) Agilent Probe ID Gene Symbol GenBank ID UniGene ID A_23_P103803 FCRH3 AF459027 Hs.292449 A_23_P104811 TREH AB000824 Hs.129712 A_23_P105100 IFITM2 X57351 Hs.174195 A_23_P107036 C17orf35 X51804 Hs.514009 A_23_P110736 C9 BC020721 Hs.1290 A_23_P111826 SPAM1 NM_003117 Hs.121494 A_23_P119533 EFNA2 AJ007292 No-Data A_23_P120105 KCNS3 BC004987 Hs.414489 A_23_P128195 HEM1 NM_005337 Hs.182014 A_23_P129332 PKD1L2 BC014157 Hs.413525 A_23_P130203 SYNGR2 AJ002308 Hs.464210 A_23_P132700 TDGF1 X14253 Hs.385870 A_23_P1331 COL13A1 NM_005203 Hs.211933 A_23_P138125 TOSO BC006401 Hs.58831 A_23_P142830 PLA2R1 U17033 Hs.410477 A_23_P146506 GOLPH2 AF236056 Hs.494337 A_23_P149569 MG29 No-Data No-Data A_23_P150590 SLC22A9 NM_080866 Hs.502772 A_23_P151166 MGC15619 BC009731 Hs.334637 A_23_P152620 TNFSF13 NM_172089 Hs.54673 A_23_P153986 KCNJ3 U39196 No-Data A_23_P154855 KCNE1 NM_000219 Hs.121495 A_23_P157380 KCTD7 AK056631 Hs.520914 A_23_P157428 SLC10A5 AK095808 Hs.531449 A_23_P160159 SLC2A5 BC001820 Hs.530003 A_23_P162162 KCTD14 NM_023930 Hs.17296 A_23_P162668 CPM BC022276 Hs.334873 A_23_P164341 VAMP2 AL050223 Hs.25348 A_23_P165394 SLC30A6 NM_017964 Hs.552598 A_23_P167276 PAQR3 AK055774 Hs.368305 A_23_P170636 KCNH8 AY053503 Hs.475656 A_23_P170736 MMP17 NM_016155 Hs.159581 A_23_P170959 LMLN NM_033029 Hs.518540 A_23_P19042 GABRB2 NM_021911 Hs.87083 A_23_P200361 CLCN6 X83378 Hs.193043 A_23_P200710 PIK3C2B -

Amino Acid Sequences Directed Against Cxcr4 And
(19) TZZ ¥¥_T (11) EP 2 285 833 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C07K 16/28 (2006.01) A61K 39/395 (2006.01) 17.12.2014 Bulletin 2014/51 A61P 31/18 (2006.01) A61P 35/00 (2006.01) (21) Application number: 09745851.7 (86) International application number: PCT/EP2009/056026 (22) Date of filing: 18.05.2009 (87) International publication number: WO 2009/138519 (19.11.2009 Gazette 2009/47) (54) AMINO ACID SEQUENCES DIRECTED AGAINST CXCR4 AND OTHER GPCRs AND COMPOUNDS COMPRISING THE SAME GEGEN CXCR4 UND ANDERE GPCR GERICHTETE AMINOSÄURESEQUENZEN SOWIE VERBINDUNGEN DAMIT SÉQUENCES D’ACIDES AMINÉS DIRIGÉES CONTRE CXCR4 ET AUTRES GPCR ET COMPOSÉS RENFERMANT CES DERNIÈRES (84) Designated Contracting States: (74) Representative: Hoffmann Eitle AT BE BG CH CY CZ DE DK EE ES FI FR GB GR Patent- und Rechtsanwälte PartmbB HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL Arabellastraße 30 PT RO SE SI SK TR 81925 München (DE) (30) Priority: 16.05.2008 US 53847 P (56) References cited: 02.10.2008 US 102142 P EP-A- 1 316 801 WO-A-99/50461 WO-A-03/050531 WO-A-03/066830 (43) Date of publication of application: WO-A-2006/089141 WO-A-2007/051063 23.02.2011 Bulletin 2011/08 • VADAY GAYLE G ET AL: "CXCR4 and CXCL12 (73) Proprietor: Ablynx N.V. (SDF-1) in prostate cancer: inhibitory effects of 9052 Ghent-Zwijnaarde (BE) human single chain Fv antibodies" CLINICAL CANCER RESEARCH, THE AMERICAN (72) Inventors: ASSOCIATION FOR CANCER RESEARCH, US, • BLANCHETOT, Christophe vol.10, no.