The Spectra of Somatic Mutations Across Many Tumor Types
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Targeting the LKB1 Tumor Suppressor
Send Orders for Reprints to [email protected] 32 Current Drug Targets, 2014, 15, 32-52 Targeting the LKB1 Tumor Suppressor Rui-Xun Zhao and Zhi-Xiang Xu* Division of Hematology and Oncology, Comprehensive Cancer Center, University of Alabama at Birmingham, Birmingham, AL, USA Abstract: LKB1 (also known as serine-threonine kinase 11, STK11) is a tumor suppressor, which is mutated or deleted in Peutz-Jeghers syndrome (PJS) and in a variety of cancers. Physiologically, LKB1 possesses multiple cellular functions in the regulation of cell bioenergetics metabolism, cell cycle arrest, embryo development, cell polarity, and apoptosis. New studies demonstrated that LKB1 may also play a role in the maintenance of function and dynamics of hematopoietic stem cells. Over the past years, personalized therapy targeting specific genetic aberrations has attracted intense interests. Within this review, several agents with potential activity against aberrant LKB1 signaling have been discussed. Potential strate- gies and challenges in targeting LKB1 inactivation are also considered. Keywords: LKB1 (serine-threonine kinase 11, STK11), AMP-activated protein kinase (AMPK), tumor suppression, mutations, targeting therapeutics. INTRODUCTION THE BIOLOGICAL FUNCTIONS OF LKB1 The LKB1 gene, also known as serine-threonine kinase Cell Metabolism 11 (STK11), was first identified by Jun-ichi Nezu of Chugai About a decade ago, studies from three different groups Pharmaceuticals in 1996 in a screen aimed at identifying new established that LKB1 is the long-sought kinase that phos- kinases [1]. The human LKB1 gene has been mapped to phorylates AMPK [9-11]. AMPK is a heterotrimeric enzyme chromosome 19p13.3. The gene spans 23 kb and is com- complex consisting of a catalytic subunit and regulatory posed of nine coding exons and a noncoding exon [2]. -

Genetic Variation Across the Human Olfactory Receptor Repertoire Alters Odor Perception
bioRxiv preprint doi: https://doi.org/10.1101/212431; this version posted November 1, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Genetic variation across the human olfactory receptor repertoire alters odor perception Casey Trimmer1,*, Andreas Keller2, Nicolle R. Murphy1, Lindsey L. Snyder1, Jason R. Willer3, Maira Nagai4,5, Nicholas Katsanis3, Leslie B. Vosshall2,6,7, Hiroaki Matsunami4,8, and Joel D. Mainland1,9 1Monell Chemical Senses Center, Philadelphia, Pennsylvania, USA 2Laboratory of Neurogenetics and Behavior, The Rockefeller University, New York, New York, USA 3Center for Human Disease Modeling, Duke University Medical Center, Durham, North Carolina, USA 4Department of Molecular Genetics and Microbiology, Duke University Medical Center, Durham, North Carolina, USA 5Department of Biochemistry, University of Sao Paulo, Sao Paulo, Brazil 6Howard Hughes Medical Institute, New York, New York, USA 7Kavli Neural Systems Institute, New York, New York, USA 8Department of Neurobiology and Duke Institute for Brain Sciences, Duke University Medical Center, Durham, North Carolina, USA 9Department of Neuroscience, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania, USA *[email protected] ABSTRACT The human olfactory receptor repertoire is characterized by an abundance of genetic variation that affects receptor response, but the perceptual effects of this variation are unclear. To address this issue, we sequenced the OR repertoire in 332 individuals and examined the relationship between genetic variation and 276 olfactory phenotypes, including the perceived intensity and pleasantness of 68 odorants at two concentrations, detection thresholds of three odorants, and general olfactory acuity. -

Overexpression and Selective Anticancer Efficacy of ENO3 in STK11 Mutant Lung Cancers
Molecules and Cells Overexpression and Selective Anticancer Efficacy of ENO3 in STK11 Mutant Lung Cancers Choa Park1,3, Yejin Lee1,3, Soyeon Je1,3, Shengzhi Chang1, Nayoung Kim1, Euna Jeong2, and Sukjoon 1,2, Yoon * 1Department of Biological Sciences, Sookmyung Women’s University, Seoul 04310, Korea, 2Research Institute of Women’s Health, Sookmyung Women’s University, Seoul 04310, Korea, 3These authors contributed equally to this work. *Correspondence: [email protected] https://doi.org/10.14348/molcells.2019.0099 www.molcells.org Oncogenic gain-of-function mutations are clinical biomarkers lung cancer, constituting ~80% to 90% of all lung can- for most targeted therapies, as well as represent direct targets cers (Novello et al., 2016; Planchard et al., 2018). NSCLC for drug treatment. Although loss-of-function mutations is classified into three types: lung adenocarcinoma (LUAD), involving the tumor suppressor gene, STK11 (LKB1) are squamous cell carcinoma, and large cell carcinoma. LUAD important in lung cancer progression, STK11 is not the direct accounts for approximately 30% of lung cancers (Gill et al., target for anticancer agents. We attempted to identify cancer 2011). Serine/threonine kinase 11 (STK11), also known as liv- transcriptome signatures associated with STK11 loss-of- er kinase B1 (LKB1), is a major tumor suppressor gene in lung function mutations. Several new sensitive and specific gene cancers. Particularly, STK11 is the most inactivated tumor expression markers (ENO3, TTC39C, LGALS3, and MAML2) suppressor gene in NSCLC (Carretero et al., 2004; Chen et were identified using two orthogonal measures, i.e., fold al., 2016; Facchinetti et al., 2017). Although STK11 inactiva- change and odds ratio analyses of transcriptome data from tion (i.e., loss-of-function mutation) occurs in ~30% of LUAD cell lines and tissue samples. -

Olfactory Receptors in Non-Chemosensory Organs: the Nervous System in Health and Disease
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Sissa Digital Library REVIEW published: 05 July 2016 doi: 10.3389/fnagi.2016.00163 Olfactory Receptors in Non-Chemosensory Organs: The Nervous System in Health and Disease Isidro Ferrer 1,2,3*, Paula Garcia-Esparcia 1,2,3, Margarita Carmona 1,2,3, Eva Carro 2,4, Eleonora Aronica 5, Gabor G. Kovacs 6, Alice Grison 7 and Stefano Gustincich 7 1 Institute of Neuropathology, Bellvitge University Hospital, Hospitalet de Llobregat, University of Barcelona, Barcelona, Spain, 2 Center for Biomedical Research in Neurodegenerative Diseases (CIBERNED), Madrid, Spain, 3 Bellvitge Biomedical Research Institute (IDIBELL), Hospitalet de Llobregat, Barcelona, Spain, 4 Neuroscience Group, Research Institute Hospital, Madrid, Spain, 5 Department of Neuropathology, Academic Medical Center, University of Amsterdam, Amsterdam, Netherlands, 6 Institute of Neurology, Medical University of Vienna, Vienna, Austria, 7 Scuola Internazionale Superiore di Studi Avanzati (SISSA), Area of Neuroscience, Trieste, Italy Olfactory receptors (ORs) and down-stream functional signaling molecules adenylyl cyclase 3 (AC3), olfactory G protein a subunit (Gaolf), OR transporters receptor transporter proteins 1 and 2 (RTP1 and RTP2), receptor expression enhancing protein 1 (REEP1), and UDP-glucuronosyltransferases (UGTs) are expressed in neurons of the human and murine central nervous system (CNS). In vitro studies have shown that these receptors react to external stimuli and therefore are equipped to be functional. However, ORs are not directly related to the detection of odors. Several molecules delivered from the blood, cerebrospinal fluid, neighboring local neurons and glial cells, distant cells through the extracellular space, and the cells’ own self-regulating internal homeostasis Edited by: Filippo Tempia, can be postulated as possible ligands. -

Incorporating Genetic Networks Into Case-Control Association Studies with High-Dimensional DNA Methylation Data Kipoong Kim and Hokeun Sun*
Kim and Sun BMC Bioinformatics (2019) 20:510 https://doi.org/10.1186/s12859-019-3040-x METHODOLOGY ARTICLE Open Access Incorporating genetic networks into case-control association studies with high-dimensional DNA methylation data Kipoong Kim and Hokeun Sun* Abstract Background: In human genetic association studies with high-dimensional gene expression data, it has been well known that statistical selection methods utilizing prior biological network knowledge such as genetic pathways and signaling pathways can outperform other methods that ignore genetic network structures in terms of true positive selection. In recent epigenetic research on case-control association studies, relatively many statistical methods have been proposed to identify cancer-related CpG sites and their corresponding genes from high-dimensional DNA methylation array data. However, most of existing methods are not designed to utilize genetic network information although methylation levels between linked genes in the genetic networks tend to be highly correlated with each other. Results: We propose new approach that combines data dimension reduction techniques with network-based regularization to identify outcome-related genes for analysis of high-dimensional DNA methylation data. In simulation studies, we demonstrated that the proposed approach overwhelms other statistical methods that do not utilize genetic network information in terms of true positive selection. We also applied it to the 450K DNA methylation array data of the four breast invasive carcinoma cancer subtypes from The Cancer Genome Atlas (TCGA) project. Conclusions: The proposed variable selection approach can utilize prior biological network information for analysis of high-dimensional DNA methylation array data. It first captures gene level signals from multiple CpG sites using data a dimension reduction technique and then performs network-based regularization based on biological network graph information. -

Qt4vh1p2c4 Nosplash E372185
Copyright 2014 by Janine Micheli-Jazdzewski ii Dedication I would like to dedicate this thesis to Rock, who is not with us anymore, TR, General Jack D. Ripper, and Page. Thank you for sitting with me while I worked for countless hours over the years. iii Acknowledgements I would like to express my special appreciation and thanks to my advisor Dr. Deanna Kroetz, you have been a superb mentor for me. I would like to thank you for encouraging my research and for helping me to grow as a research scientist. Your advice on both research, as well as on my career have been priceless. I would also like to thank my committee members, Dr. Laura Bull, Dr. Steve Hamilton and Dr. John Witte for guiding my research and expanding my knowledge on statistics, genetics and clinical phenotypes. I also want to thank past and present members of my laboratory for their support and help over the years, especially Dr. Mike Baldwin, Dr. Sveta Markova, Dr. Ying Mei Liu and Dr. Leslie Chinn. Thanks are also due to my many collaborators that made this research possible including: Dr. Eric Jorgenson, Dr. David Bangsberg, Dr. Taisei Mushiroda, Dr. Michiaki Kubo, Dr. Yusuke Nakamura, Dr. Jeffrey Martin, Joel Mefford, Dr. Sarah Shutgarts, Dr. Sulggi Lee and Dr. Sook Wah Yee. A special thank you to the RIKEN Center for Genomic Medicine that generously performed the genome-wide genotyping for these projects. Thanks to Dr. Steve Chamow, Dr. Bill Werner, Dr. Montse Carrasco, and Dr. Teresa Chen who started me on the path to becoming a scientist. -
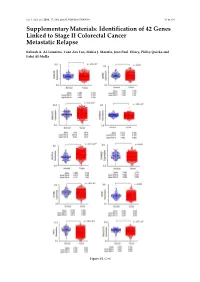
Identification of 42 Genes Linked to Stage II Colorectal Cancer Metastatic Relapse
Int. J. Mol. Sci. 2016, 17, 598; doi:10.3390/ijms17040598 S1 of S16 Supplementary Materials: Identification of 42 Genes Linked to Stage II Colorectal Cancer Metastatic Relapse Rabeah A. Al-Temaimi, Tuan Zea Tan, Makia J. Marafie, Jean Paul Thiery, Philip Quirke and Fahd Al-Mulla Figure S1. Cont. Int. J. Mol. Sci. 2016, 17, 598; doi:10.3390/ijms17040598 S2 of S16 Figure S1. Mean expression levels of fourteen genes of significant association with CRC DFS and OS that are differentially expressed in normal colon compared to CRC tissues. Each dot represents a sample. Table S1. Copy number aberrations associated with poor disease-free survival and metastasis in early stage II CRC as predicted by STAC and SPPS combined methodologies with resident gene symbols. CN stands for copy number, whereas CNV is copy number variation. Region Cytoband % of CNV Count of Region Event Gene Symbols Length Location Overlap Genes chr1:113,025,076–113,199,133 174,057 p13.2 CN Loss 0.0 2 AKR7A2P1, SLC16A1 chr1:141,465,960–141,822,265 356,305 q12–q21.1 CN Gain 95.9 1 SRGAP2B MIR5087, LOC10013000 0, FLJ39739, LOC10028679 3, PPIAL4G, PPIAL4A, NBPF14, chr1:144,911,564–146,242,907 1,331,343 q21.1 CN Gain 99.6 16 NBPF15, NBPF16, PPIAL4E, NBPF16, PPIAL4D, PPIAL4F, LOC645166, LOC388692, FCGR1C chr1:177,209,428–177,226,812 17,384 q25.3 CN Gain 0.0 0 chr1:197,652,888–197,676,831 23,943 q32.1 CN Gain 0.0 1 KIF21B chr1:201,015,278–201,033,308 18,030 q32.1 CN Gain 0.0 1 PLEKHA6 chr1:201,289,154–201,298,247 9093 q32.1 CN Gain 0.0 0 chr1:216,820,186–217,043,421 223,235 q41 CN -

Identification of Candidate Biomarkers and Pathways Associated with Type 1 Diabetes Mellitus Using Bioinformatics Analysis
bioRxiv preprint doi: https://doi.org/10.1101/2021.06.08.447531; this version posted June 9, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Identification of candidate biomarkers and pathways associated with type 1 diabetes mellitus using bioinformatics analysis Basavaraj Vastrad1, Chanabasayya Vastrad*2 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karnataka, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2021.06.08.447531; this version posted June 9, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Type 1 diabetes mellitus (T1DM) is a metabolic disorder for which the underlying molecular mechanisms remain largely unclear. This investigation aimed to elucidate essential candidate genes and pathways in T1DM by integrated bioinformatics analysis. In this study, differentially expressed genes (DEGs) were analyzed using DESeq2 of R package from GSE162689 of the Gene Expression Omnibus (GEO). Gene ontology (GO) enrichment analysis, REACTOME pathway enrichment analysis, and construction and analysis of protein-protein interaction (PPI) network, modules, miRNA-hub gene regulatory network and TF-hub gene regulatory network, and validation of hub genes were then performed. A total of 952 DEGs (477 up regulated and 475 down regulated genes) were identified in T1DM. GO and REACTOME enrichment result results showed that DEGs mainly enriched in multicellular organism development, detection of stimulus, diseases of signal transduction by growth factor receptors and second messengers, and olfactory signaling pathway. -

WO 2019/068007 Al Figure 2
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization I International Bureau (10) International Publication Number (43) International Publication Date WO 2019/068007 Al 04 April 2019 (04.04.2019) W 1P O PCT (51) International Patent Classification: (72) Inventors; and C12N 15/10 (2006.01) C07K 16/28 (2006.01) (71) Applicants: GROSS, Gideon [EVIL]; IE-1-5 Address C12N 5/10 (2006.0 1) C12Q 1/6809 (20 18.0 1) M.P. Korazim, 1292200 Moshav Almagor (IL). GIBSON, C07K 14/705 (2006.01) A61P 35/00 (2006.01) Will [US/US]; c/o ImmPACT-Bio Ltd., 2 Ilian Ramon St., C07K 14/725 (2006.01) P.O. Box 4044, 7403635 Ness Ziona (TL). DAHARY, Dvir [EilL]; c/o ImmPACT-Bio Ltd., 2 Ilian Ramon St., P.O. (21) International Application Number: Box 4044, 7403635 Ness Ziona (IL). BEIMAN, Merav PCT/US2018/053583 [EilL]; c/o ImmPACT-Bio Ltd., 2 Ilian Ramon St., P.O. (22) International Filing Date: Box 4044, 7403635 Ness Ziona (E.). 28 September 2018 (28.09.2018) (74) Agent: MACDOUGALL, Christina, A. et al; Morgan, (25) Filing Language: English Lewis & Bockius LLP, One Market, Spear Tower, SanFran- cisco, CA 94105 (US). (26) Publication Language: English (81) Designated States (unless otherwise indicated, for every (30) Priority Data: kind of national protection available): AE, AG, AL, AM, 62/564,454 28 September 2017 (28.09.2017) US AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, 62/649,429 28 March 2018 (28.03.2018) US CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, (71) Applicant: IMMP ACT-BIO LTD. -
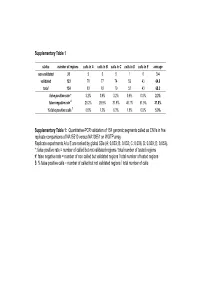
Quantitative-PCR Validation of 154 Genomic Segments Called As Cnvs in Five Replicat
Supplementary Table 1 status number of regions calls in A calls in B calls in C calls in D calls in E average non validated 31 5 6 5 1 0 3.4 validated 123 78 77 74 52 43 64.8 total 154 83 83 79 53 43 68.2 false positive rate * 3.2% 3.9% 3.2% 0.6% 0.0% 2.2% false negative rate # 29.2% 29.9% 31.8% 46.1% 51.9% 37.8% % false positive calls $ 6.0% 7.2% 6.3% 1.9% 0.0% 5.0% Supplementary Table 1: Quantitative-PCR validation of 154 genomic segments called as CNVs in five replicate comparisons of NA15510 versus NA10851 on WGTP array Replicate experiments A to E are ranked by global SDe (A: 0.033; B: 0.033; C: 0.036; D: 0.039; E: 0.053). *: false positive rate = number of called but not validated regions / total number of tested regions #: false negative rate = number of non called but validated regions / total number of tested regions $: % false positive calls = number of called but not validated regions / total number of calls False positive estimates for 500K EA CNV calls Total Rep1 Rep2 Rep3 Avg (unique) Validated 33 28 32 31 38 Not validated 2 2 2 2 5 Total 35 30 34 33 43 % False positive 5.71% 6.67% 5.88% 6.09% - % False negative 13.16% 26.32% 15.79% 18.42% - Supplementary Table 2A : Quantitative PCR validation of 43 unique CNV regions called as CNVs in three replicate comparisons of NA15510 versus NA10851 using the 500K EA array. -

Clinical, Molecular, and Immune Analysis of Dabrafenib-Trametinib
Supplementary Online Content Chen G, McQuade JL, Panka DJ, et al. Clinical, molecular and immune analysis of dabrafenib-trametinib combination treatment for metastatic melanoma that progressed during BRAF inhibitor monotherapy: a phase 2 clinical trial. JAMA Oncology. Published online April 28, 2016. doi:10.1001/jamaoncol.2016.0509. eMethods. eReferences. eTable 1. Clinical efficacy eTable 2. Adverse events eTable 3. Correlation of baseline patient characteristics with treatment outcomes eTable 4. Patient responses and baseline IHC results eFigure 1. Kaplan-Meier analysis of overall survival eFigure 2. Correlation between IHC and RNAseq results eFigure 3. pPRAS40 expression and PFS eFigure 4. Baseline and treatment-induced changes in immune infiltrates eFigure 5. PD-L1 expression eTable 5. Nonsynonymous mutations detected by WES in baseline tumors This supplementary material has been provided by the authors to give readers additional information about their work. © 2016 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 09/30/2021 eMethods Whole exome sequencing Whole exome capture libraries for both tumor and normal samples were constructed using 100ng genomic DNA input and following the protocol as described by Fisher et al.,3 with the following adapter modification: Illumina paired end adapters were replaced with palindromic forked adapters with unique 8 base index sequences embedded within the adapter. In-solution hybrid selection was performed using the Illumina Rapid Capture Exome enrichment kit with 38Mb target territory (29Mb baited). The targeted region includes 98.3% of the intervals in the Refseq exome database. Dual-indexed libraries were pooled into groups of up to 96 samples prior to hybridization. -

1 Copper-Mediated Thiol Potentiation and Mutagenesis-Guided Modeling Suggest a Highly Conserved Copper Binding Motif in Human OR
Copper-mediated thiol potentiation and mutagenesis-guided modeling suggest a highly conserved copper binding motif in human OR2M3 Franziska Haag1, Lucky Ahmed2, Krystle Reiss2, Eric Block3, Victor S. Batista2 and Dietmar Krautwurst1 1Leibniz-Institute for Food Systems Biology at the Technical University of Munich, Lise-Meitner-Str. 34, D-85354 Freising, Germany 2Department of Chemistry, Yale University, New Haven, CT 06520, United States 3Department of Chemistry, University at Albany, State University of New York, Albany, NY 12222, United States Contents: Table S1: Oligonucleotides for molecular cloning of odorant receptors investigated. ........................... 2 Table S2: Oligonucleotides for Homo sapiens OR2M3 site-directed mutagenesis. ................................ 2 Table S3: Oligonucleotides for Homo sapiens OR2W1 site-directed mutagenesis. ................................ 4 Table S4: Vector internal oligonucleotides for pi2-dk (39aa rho-tag). .................................................... 4 Table S5: NCBI reference sequences of olfactory receptor genes investigated. ..................................... 5 Table S6: EC50 values and relative amplitudes for 3-mercapto-2-methylpentan-1-ol on OR2M3 wild type in the absence and presence of different heavy metals. ................................................................ 7 Table S7: EC50 values and relative amplitudes for OR2M3 wild type in the absence and presence of different concentrations of Cu2+. ...........................................................................................................