Prognostic Classifier Based on Genome-Wide DNA Methylation Profiling in Well-Differentiated Thyroid Tumors
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Whole Exome Sequencing in Families at High Risk for Hodgkin Lymphoma: Identification of a Predisposing Mutation in the KDR Gene
Hodgkin Lymphoma SUPPLEMENTARY APPENDIX Whole exome sequencing in families at high risk for Hodgkin lymphoma: identification of a predisposing mutation in the KDR gene Melissa Rotunno, 1 Mary L. McMaster, 1 Joseph Boland, 2 Sara Bass, 2 Xijun Zhang, 2 Laurie Burdett, 2 Belynda Hicks, 2 Sarangan Ravichandran, 3 Brian T. Luke, 3 Meredith Yeager, 2 Laura Fontaine, 4 Paula L. Hyland, 1 Alisa M. Goldstein, 1 NCI DCEG Cancer Sequencing Working Group, NCI DCEG Cancer Genomics Research Laboratory, Stephen J. Chanock, 5 Neil E. Caporaso, 1 Margaret A. Tucker, 6 and Lynn R. Goldin 1 1Genetic Epidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 2Cancer Genomics Research Laboratory, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 3Ad - vanced Biomedical Computing Center, Leidos Biomedical Research Inc.; Frederick National Laboratory for Cancer Research, Frederick, MD; 4Westat, Inc., Rockville MD; 5Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; and 6Human Genetics Program, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD, USA ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2015.135475 Received: August 19, 2015. Accepted: January 7, 2016. Pre-published: June 13, 2016. Correspondence: [email protected] Supplemental Author Information: NCI DCEG Cancer Sequencing Working Group: Mark H. Greene, Allan Hildesheim, Nan Hu, Maria Theresa Landi, Jennifer Loud, Phuong Mai, Lisa Mirabello, Lindsay Morton, Dilys Parry, Anand Pathak, Douglas R. Stewart, Philip R. Taylor, Geoffrey S. Tobias, Xiaohong R. Yang, Guoqin Yu NCI DCEG Cancer Genomics Research Laboratory: Salma Chowdhury, Michael Cullen, Casey Dagnall, Herbert Higson, Amy A. -

Incorporating Genetic Networks Into Case-Control Association Studies with High-Dimensional DNA Methylation Data Kipoong Kim and Hokeun Sun*
Kim and Sun BMC Bioinformatics (2019) 20:510 https://doi.org/10.1186/s12859-019-3040-x METHODOLOGY ARTICLE Open Access Incorporating genetic networks into case-control association studies with high-dimensional DNA methylation data Kipoong Kim and Hokeun Sun* Abstract Background: In human genetic association studies with high-dimensional gene expression data, it has been well known that statistical selection methods utilizing prior biological network knowledge such as genetic pathways and signaling pathways can outperform other methods that ignore genetic network structures in terms of true positive selection. In recent epigenetic research on case-control association studies, relatively many statistical methods have been proposed to identify cancer-related CpG sites and their corresponding genes from high-dimensional DNA methylation array data. However, most of existing methods are not designed to utilize genetic network information although methylation levels between linked genes in the genetic networks tend to be highly correlated with each other. Results: We propose new approach that combines data dimension reduction techniques with network-based regularization to identify outcome-related genes for analysis of high-dimensional DNA methylation data. In simulation studies, we demonstrated that the proposed approach overwhelms other statistical methods that do not utilize genetic network information in terms of true positive selection. We also applied it to the 450K DNA methylation array data of the four breast invasive carcinoma cancer subtypes from The Cancer Genome Atlas (TCGA) project. Conclusions: The proposed variable selection approach can utilize prior biological network information for analysis of high-dimensional DNA methylation array data. It first captures gene level signals from multiple CpG sites using data a dimension reduction technique and then performs network-based regularization based on biological network graph information. -

1 Genome-Wide Comparisons of Variation in Linkage Disequilibrium
Downloaded from genome.cshlp.org on September 30, 2021 - Published by Cold Spring Harbor Laboratory Press Genome-wide comparisons of variation in linkage disequilibrium Yik Y. Teo1,*, Andrew E. Fry1, Kanishka Bhattacharya1, Kerrin S. Small1, Dominic P. Kwiatkowski1,2, Taane G. Clark1,2 1 Wellcome Trust Centre for Human Genetics, University of Oxford, United Kingdom 2 Wellcome Trust Sanger Institute, Hinxton, United Kingdom Running title: Genome-wide comparisons of LD Keywords: linkage disequilibrium, imputation, positive selection, meta-analysis, genome-wide association study * Corresponding author: Wellcome Trust Centre for Human Genetics, Roosevelt Drive, Oxford OX3 7BN, United Kingdom. Email: [email protected], phone: +44 1865 287712, fax: +44 1865 287 501. ABSTRACT Current genome-wide surveys of common diseases and complex traits fundamentally aim to detect indirect associations where the SNPs carrying the association signals are not biologically active but are in linkage disequilibrium (LD) with some unknown functional polymorphisms. Reproducing any novel discoveries from these genome-wide scans in independent studies is now a prerequisite for the putative findings to be accepted. Significant differences in patterns of LD between populations can affect the portability of phenotypic associations when the replication effort or meta-analyses are attempted in populations that are distinct from the original population which the genome-wide study is performed in. Here we introduce a novel method for genome-wide analyses of LD variations between populations that allow the identification of candidate regions with different patterns of LD. The evidence of LD variation provided by the introduced method correlated with the degree of differences in the frequencies of the most common haplotype across the populations. -

Molecular Correlates of Metastasis by Systematic Pan-Cancer Analysis Across the Cancer Genome Atlas
Author Manuscript Published OnlineFirst on November 6, 2018; DOI: 10.1158/1541-7786.MCR-18-0601 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Molecular correlates of metastasis by systematic pan-cancer analysis across The Cancer Genome Atlas Fengju Chen1*, Yiqun Zhang1*, Sooryanarayana Varambally2,3, Chad J. Creighton1,4,5,6 1. Dan L. Duncan Comprehensive Cancer Center Division of Biostatistics, Baylor College of Medicine, Houston, TX, USA. 2. Comprehensive Cancer Center, University of Alabama at Birmingham, Birmingham, AL 35233, USA 3. Molecular and Cellular Pathology, Department of Pathology, University of Alabama at Birmingham, Birmingham, AL 35233, USA 4. Department of Bioinformatics and Computational Biology, The University of Texas MD Anderson Cancer Center, Houston, TX, USA 5. Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX 77030, USA 6. Department of Medicine, Baylor College of Medicine, Houston, TX, USA * co-first authors Running title: Pan-cancer metastasis correlates Abbreviations: TCGA, The Cancer Genome Atlas; RNA-seq, RNA sequencing; RPPA, reverse- phase protein arrays Correspondence to: Chad J. Creighton ([email protected]) One Baylor Plaza, MS305 Houston, TX 77030 Disclosure of potential conflicts of interest: The authors have no conflicts of interest. 1 Downloaded from mcr.aacrjournals.org on September 28, 2021. © 2018 American Association for Cancer Research. Author Manuscript Published OnlineFirst on November 6, 2018; DOI: 10.1158/1541-7786.MCR-18-0601 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Abstract Tumor metastasis is a major contributor to cancer patient mortality, but the process remains poorly understood. -

Durbinharly.Pdf (9.770Mb)
GENOMICS OF SEASONAL HAIR SHEDDING AND ECOREGION-SPECIFIC GROWTH TO IDENTIFY ENVIRONMENTALLY-ADAPTED BEEF CATTLE _______________________________________ A Dissertation presented to the Faculty of the Graduate School at the University of Missouri-Columbia _______________________________________________________ In Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy _____________________________________________________ by HARLY JANE DURBIN Dr. Jared Decker, Dissertation Supervisor December 2020 APPROVAL PAGE The undersigned, appointed by the Dean of the Graduate School, have examined the dissertation entitled: GENOMICS OF SEASONAL HAIR SHEDDING AND ECO-REGION SPECIFIC GROWTH TO IDENTIFY ENVIRONMENTALLY-ADAPTED BEEF CATTLE Presented by Harly Jane Durbin, a candidate for the degree of Doctor of Philosophy, and hereby certify that in their opinion it is worthy of acceptance. _____________________________________ Dr. Jared E. Decker, Animal Sciences, UMC _____________________________________ Dr. Robert D. Schnabel, Animal Sciences, UMC _____________________________________ Dr. Jeremy F. Taylor, Animal Sciences, UMC _____________________________________ Dr. Elizabeth G. King, Biological Sciences, UMC _____________________________________ Dr. Stephen P. Miller, Angus Genetics, Inc. DEDICATION Momma – thank you for teaching me compassion and also how to approach life with a sense of humor. You do such an incredible amount for other people and I strive to be as selfless every day. Daddy – thank you for encouraging me to think independently and for teaching me how to stand up for myself. Thank you also for the years of FFA (also known sometimes as Father Feeds Animal) and getting up early to haul heifers to jackpot shows. Darby – thank you for holding me to a high standard. I often think there was some mix-up and that you were meant to be the older sister for how much you hold me up. -

9-Marie-Francoise Ritz MS
Send Orders for Reprints to [email protected] 478 Current Neurovascular Research, 2019, 16, 478-490 RESEARCH ARTICLE Combined Transcriptomic and Proteomic Analyses of Cerebral Frontal Lobe Tissue Identified RNA Metabolism Dysregulation as One Potential Pathogenic Mechanism in Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL) Marie-Françoise Ritz1,*, Paul Jenoe2, Leo Bonati3, Stefan Engelter3,4, Philippe Lyrer3 and Nils Peters3,4 1Department of Biomedicine, Brain Tumor Biology Laboratory, University of Basel, and University Hospital of Basel, Hebelstrasse 20, 4031 Basel, Switzerland; 2Proteomics Core Facility, Biocenter, University of Basel, Klingelbergstrasse 50/70, 4056 Basel, Switzerland; 3Department of Neurology and Stroke Center, University Hospital Basel and University of Basel, Petersgraben 4, 4031 Basel, Switzerland; 4Neurorehabilitation Unit, University of Basel and University Center for Medicine of Aging, Felix Platter Hospital, Burgfelderstrasse 101, 4055 Basel, Switzerland Abstract: Background: Cerebral small vessel disease (SVD) is an important cause of stroke and vascular cognitive impairment (VCI), leading to subcortical ischemic vascular dementia. As a he- reditary form of SVD with early onset, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) represents a pure form of SVD and may thus serve as a model disease for SVD. To date, underlying molecular mechanisms linking vascular pathol- ogy and subsequent neuronal damage in SVD are incompletely understood. A R T I C L E H I S T O R Y Objective: We performed comparative transcriptional profiling microarray and proteomic analyses Received: October 01, 2019 on post-mortem frontal lobe specimen from 2 CADASIL patients and 5 non neurologically dis- Revised: October 11, 2019 Accepted: October 15, 2019 eased controls in order to identify dysregulated pathways potentially involved in the development DOI: of tissue damage in CADASIL. -

Table S3. RAE Analysis of Well-Differentiated Liposarcoma
Table S3. RAE analysis of well-differentiated liposarcoma Model Chromosome Region start Region end Size q value freqX0* # genes Genes Amp 1 145009467 145122002 112536 0.097 21.8 2 PRKAB2,PDIA3P Amp 1 145224467 146188434 963968 0.029 23.6 10 CHD1L,BCL9,ACP6,GJA5,GJA8,GPR89B,GPR89C,PDZK1P1,RP11-94I2.2,NBPF11 Amp 1 147475854 148412469 936616 0.034 23.6 20 PPIAL4A,FCGR1A,HIST2H2BF,HIST2H3D,HIST2H2AA4,HIST2H2AA3,HIST2H3A,HIST2H3C,HIST2H4B,HIST2H4A,HIST2H2BE, HIST2H2AC,HIST2H2AB,BOLA1,SV2A,SF3B4,MTMR11,OTUD7B,VPS45,PLEKHO1 Amp 1 148582896 153398462 4815567 1.5E-05 49.1 152 PRPF3,RPRD2,TARS2,ECM1,ADAMTSL4,MCL1,ENSA,GOLPH3L,HORMAD1,CTSS,CTSK,ARNT,SETDB1,LASS2,ANXA9, FAM63A,PRUNE,BNIPL,C1orf56,CDC42SE1,MLLT11,GABPB2,SEMA6C,TNFAIP8L2,LYSMD1,SCNM1,TMOD4,VPS72, PIP5K1A,PSMD4,ZNF687,PI4KB,RFX5,SELENBP1,PSMB4,POGZ,CGN,TUFT1,SNX27,TNRC4,MRPL9,OAZ3,TDRKH,LINGO4, RORC,THEM5,THEM4,S100A10,S100A11,TCHHL1,TCHH,RPTN,HRNR,FLG,FLG2,CRNN,LCE5A,CRCT1,LCE3E,LCE3D,LCE3C,LCE3B, LCE3A,LCE2D,LCE2C,LCE2B,LCE2A,LCE4A,KPRP,LCE1F,LCE1E,LCE1D,LCE1C,LCE1B,LCE1A,SMCP,IVL,SPRR4,SPRR1A,SPRR3, SPRR1B,SPRR2D,SPRR2A,SPRR2B,SPRR2E,SPRR2F,SPRR2C,SPRR2G,LELP1,LOR,PGLYRP3,PGLYRP4,S100A9,S100A12,S100A8, S100A7A,S100A7L2,S100A7,S100A6,S100A5,S100A4,S100A3,S100A2,S100A16,S100A14,S100A13,S100A1,C1orf77,SNAPIN,ILF2, NPR1,INTS3,SLC27A3,GATAD2B,DENND4B,CRTC2,SLC39A1,CREB3L4,JTB,RAB13,RPS27,NUP210L,TPM3,C1orf189,C1orf43,UBAP2L,HAX1, AQP10,ATP8B2,IL6R,SHE,TDRD10,UBE2Q1,CHRNB2,ADAR,KCNN3,PMVK,PBXIP1,PYGO2,SHC1,CKS1B,FLAD1,LENEP,ZBTB7B,DCST2, DCST1,ADAM15,EFNA4,EFNA3,EFNA1,RAG1AP1,DPM3 Amp 1 -

1 Copper-Mediated Thiol Potentiation and Mutagenesis-Guided Modeling Suggest a Highly Conserved Copper Binding Motif in Human OR
Copper-mediated thiol potentiation and mutagenesis-guided modeling suggest a highly conserved copper binding motif in human OR2M3 Franziska Haag1, Lucky Ahmed2, Krystle Reiss2, Eric Block3, Victor S. Batista2 and Dietmar Krautwurst1 1Leibniz-Institute for Food Systems Biology at the Technical University of Munich, Lise-Meitner-Str. 34, D-85354 Freising, Germany 2Department of Chemistry, Yale University, New Haven, CT 06520, United States 3Department of Chemistry, University at Albany, State University of New York, Albany, NY 12222, United States Contents: Table S1: Oligonucleotides for molecular cloning of odorant receptors investigated. ........................... 2 Table S2: Oligonucleotides for Homo sapiens OR2M3 site-directed mutagenesis. ................................ 2 Table S3: Oligonucleotides for Homo sapiens OR2W1 site-directed mutagenesis. ................................ 4 Table S4: Vector internal oligonucleotides for pi2-dk (39aa rho-tag). .................................................... 4 Table S5: NCBI reference sequences of olfactory receptor genes investigated. ..................................... 5 Table S6: EC50 values and relative amplitudes for 3-mercapto-2-methylpentan-1-ol on OR2M3 wild type in the absence and presence of different heavy metals. ................................................................ 7 Table S7: EC50 values and relative amplitudes for OR2M3 wild type in the absence and presence of different concentrations of Cu2+. ........................................................................................................... -

An Analysis About Heterogeneity Among Cancers Based on the DNA Methylation Patterns
An analysis about heterogeneity among cancers based on the DNA Methylation Patterns Yang Liu Harbin Institute of Technology Yue Gu Harbin Medical University Mu Su Harbin Institute of Technology Hui Liu Harbin Institute of Technology Shumei Zhang Harbin Medical University Yan Zhang ( [email protected] ) Harbin Institute of Technology https://orcid.org/0000-0002-5307-2484 Research article Keywords: DNA methylation, cancer, epigenetic heterogeneity, survival analysis Posted Date: July 17th, 2019 DOI: https://doi.org/10.21203/rs.2.11636/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Version of Record: A version of this preprint was published at BMC Cancer on December 1st, 2019. See the published version at https://doi.org/10.1186/s12885-019-6455-x. Page 1/28 Abstract Background: The occurrence of cancer is usually the result of a co-effect of genetic and environmental factors. It is generally believed that the main cause of cancer is the accumulation of genetic mutations, and DNA methylation, as one of the epigenetic modications closely related to environmental factors, participates in the regulation of gene expression and cell differentiation and plays an important role in the development of cancer. Methods: This article discusses the epigenetic heterogeneity of cancer in detail. Firstly DNA methylation data of 7 cancer types were obtained from Illumina Innium HumanMethylation 450K platform of TCGA database. Diagnostic markers of each cancer were obtained by t-test and absolute difference of DNA differencial methylation analysis. Enrichment analysis of these specic markers indicated that they were involved in different biological functions. -

Strong Selection During the Last Millennium for African Ancestry In
Strong selection during the last millennium for African ancestry in the admixed population of Madagascar Denis Pierron, Margit Heiske, Harilanto Razafindrazaka, Veronica Pereda-Loth, Jazmin Sanchez, Omar Alva, Amal Arachiche, Anne Boland, Robert Olaso, Jean-François Deleuze, et al. To cite this version: Denis Pierron, Margit Heiske, Harilanto Razafindrazaka, Veronica Pereda-Loth, Jazmin Sanchez, et al.. Strong selection during the last millennium for African ancestry in the admixed population of Mada- gascar. Nature Communications, Nature Publishing Group, 2018, 9 (1), pp.932. 10.1038/s41467-018- 03342-5. hal-02112693 HAL Id: hal-02112693 https://hal.archives-ouvertes.fr/hal-02112693 Submitted on 9 Jan 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License ARTICLE DOI: 10.1038/s41467-018-03342-5 OPEN Strong selection during the last millennium for African ancestry in the admixed population of Madagascar Denis Pierron1, Margit Heiske1, Harilanto Razafindrazaka2,1, Veronica Pereda-loth1, Jazmin Sanchez1, Omar Alva1, Amal Arachiche1, Anne Boland3, Robert Olaso3, Jean-Francois Deleuze3, Francois-Xavier Ricaut1, Jean-Aimé Rakotoarisoa4, Chantal Radimilahy4, Mark Stoneking5 & Thierry Letellier1 1234567890():,; While admixed populations offer a unique opportunity to detect selection, the admixture in most of the studied populations occurred too recently to produce conclusive signals. -
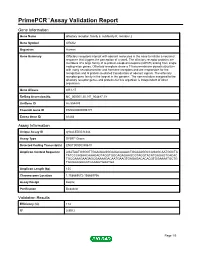
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name olfactory receptor, family 6, subfamily K, member 2 Gene Symbol OR6K2 Organism Human Gene Summary Olfactory receptors interact with odorant molecules in the nose to initiate a neuronal response that triggers the perception of a smell. The olfactory receptor proteins are members of a large family of G-protein-coupled receptors (GPCR) arising from single coding-exon genes. Olfactory receptors share a 7-transmembrane domain structure with many neurotransmitter and hormone receptors and are responsible for the recognition and G protein-mediated transduction of odorant signals. The olfactory receptor gene family is the largest in the genome. The nomenclature assigned to the olfactory receptor genes and proteins for this organism is independent of other organisms. Gene Aliases OR1-17 RefSeq Accession No. NC_000001.10, NT_004487.19 UniGene ID Hs.554489 Ensembl Gene ID ENSG00000196171 Entrez Gene ID 81448 Assay Information Unique Assay ID qHsaCED0034344 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000359610 Amplicon Context Sequence AGATAATGGGGTTGAAGAAGGGAGACAAAACTGCAAAGGCCAGAGCAATGGCTA TATCCCAGAACAAAGAGTAGGTGGCAGAGAAGCGTAGGTACATGAGAGTCACAC TGCCAAAGAAGAGCGAAAAGACAATGAAGTGAGAGACACACGTGGAAAATGCTG TGCGGCGGCCTCCAGCTGAATGA Amplicon Length (bp) 155 Chromosome Location 1:158669572-158669756 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 114 R2 0.9912 Page 1/5 PrimePCR™Assay Validation Report cDNA Cq 33.55 cDNA Tm (Celsius) 83 gDNA Cq 23.81 Specificity -

The Hypothalamus As a Hub for SARS-Cov-2 Brain Infection and Pathogenesis
bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.139329; this version posted June 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. The hypothalamus as a hub for SARS-CoV-2 brain infection and pathogenesis Sreekala Nampoothiri1,2#, Florent Sauve1,2#, Gaëtan Ternier1,2ƒ, Daniela Fernandois1,2 ƒ, Caio Coelho1,2, Monica ImBernon1,2, Eleonora Deligia1,2, Romain PerBet1, Vincent Florent1,2,3, Marc Baroncini1,2, Florence Pasquier1,4, François Trottein5, Claude-Alain Maurage1,2, Virginie Mattot1,2‡, Paolo GiacoBini1,2‡, S. Rasika1,2‡*, Vincent Prevot1,2‡* 1 Univ. Lille, Inserm, CHU Lille, Lille Neuroscience & Cognition, DistAlz, UMR-S 1172, Lille, France 2 LaBoratorY of Development and PlasticitY of the Neuroendocrine Brain, FHU 1000 daYs for health, EGID, School of Medicine, Lille, France 3 Nutrition, Arras General Hospital, Arras, France 4 Centre mémoire ressources et recherche, CHU Lille, LiCEND, Lille, France 5 Univ. Lille, CNRS, INSERM, CHU Lille, Institut Pasteur de Lille, U1019 - UMR 8204 - CIIL - Center for Infection and ImmunitY of Lille (CIIL), Lille, France. # and ƒ These authors contriButed equallY to this work. ‡ These authors directed this work *Correspondence to: [email protected] and [email protected] Short title: Covid-19: the hypothalamic hypothesis 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.139329; this version posted June 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.