Microarray-Based Analysis of Genes, Transcription Factors, and Epigenetic Modifications in Lung Cancer Exposed to Nitric Oxide
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Data
Figure 2S 4 7 A - C 080125 CSCs 080418 CSCs - + IFN-a 48 h + IFN-a 48 h + IFN-a 72 h 6 + IFN-a 72 h 3 5 MRFI 4 2 3 2 1 1 0 0 MHC I MHC II MICA MICB ULBP-1 ULBP-2 ULBP-3 ULBP-4 MHC I MHC II MICA MICB ULBP-1 ULBP-2 ULBP-3 ULBP-4 7 B 13 080125 FBS - D 080418 FBS - + IFN-a 48 h 12 + IFN-a 48 h + IFN-a 72 h + IFN-a 72 h 6 080125 FBS 11 10 5 9 8 4 7 6 3 MRFI 5 4 2 3 2 1 1 0 0 MHC I MHC II MICA MICB ULBP-1 ULBP-2 ULBP-3 ULBP-4 MHC I MHC II MICA MICB ULBP-1 ULBP-2 ULBP-3 ULBP-4 Molecule Molecule FIGURE 4S FIGURE 5S Panel A Panel B FIGURE 6S A B C D Supplemental Results Table 1S. Modulation by IFN-α of APM in GBM CSC and FBS tumor cell lines. Molecule * Cell line IFN-α‡ HLA β2-m# HLA LMP TAP1 TAP2 class II A A HC§ 2 7 10 080125 CSCs - 1∞ (1) 3 (65) 2 (91) 1 (2) 6 (47) 2 (61) 1 (3) 1 (2) 1 (3) + 2 (81) 11 (80) 13 (99) 1 (3) 8 (88) 4 (91) 1 (2) 1 (3) 2 (68) 080125 FBS - 2 (81) 4 (63) 4 (83) 1 (3) 6 (80) 3 (67) 2 (86) 1 (3) 2 (75) + 2 (99) 14 (90) 7 (97) 5 (75) 7 (100) 6 (98) 2 (90) 1 (4) 3 (87) 080418 CSCs - 2 (51) 1 (1) 1 (3) 2 (47) 2 (83) 2 (54) 1 (4) 1 (2) 1 (3) + 2 (81) 3 (76) 5 (75) 2 (50) 2 (83) 3 (71) 1 (3) 2 (87) 1 (2) 080418 FBS - 1 (3) 3 (70) 2 (88) 1 (4) 3 (87) 2 (76) 1 (3) 1 (3) 1 (2) + 2 (78) 7 (98) 5 (99) 2 (94) 5 (100) 3 (100) 1 (4) 2 (100) 1 (2) 070104 CSCs - 1 (2) 1 (3) 1 (3) 2 (78) 1 (3) 1 (2) 1 (3) 1 (3) 1 (2) + 2 (98) 8 (100) 10 (88) 4 (89) 3 (98) 3 (94) 1 (4) 2 (86) 2 (79) * expression of APM molecules was evaluated by intracellular staining and cytofluorimetric analysis; ‡ cells were treatead or not (+/-) for 72 h with 1000 IU/ml of IFN-α; # β-2 microglobulin; § β-2 microglobulin-free HLA-A heavy chain; ∞ values are indicated as ratio between the mean of fluorescence intensity of cells stained with the selected mAb and that of the negative control; bold values indicate significant MRFI (≥ 2). -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Supplemental Materials ZNF281 Enhances Cardiac Reprogramming
Supplemental Materials ZNF281 enhances cardiac reprogramming by modulating cardiac and inflammatory gene expression Huanyu Zhou, Maria Gabriela Morales, Hisayuki Hashimoto, Matthew E. Dickson, Kunhua Song, Wenduo Ye, Min S. Kim, Hanspeter Niederstrasser, Zhaoning Wang, Beibei Chen, Bruce A. Posner, Rhonda Bassel-Duby and Eric N. Olson Supplemental Table 1; related to Figure 1. Supplemental Table 2; related to Figure 1. Supplemental Table 3; related to the “quantitative mRNA measurement” in Materials and Methods section. Supplemental Table 4; related to the “ChIP-seq, gene ontology and pathway analysis” and “RNA-seq” and gene ontology analysis” in Materials and Methods section. Supplemental Figure S1; related to Figure 1. Supplemental Figure S2; related to Figure 2. Supplemental Figure S3; related to Figure 3. Supplemental Figure S4; related to Figure 4. Supplemental Figure S5; related to Figure 6. Supplemental Table S1. Genes included in human retroviral ORF cDNA library. Gene Gene Gene Gene Gene Gene Gene Gene Symbol Symbol Symbol Symbol Symbol Symbol Symbol Symbol AATF BMP8A CEBPE CTNNB1 ESR2 GDF3 HOXA5 IL17D ADIPOQ BRPF1 CEBPG CUX1 ESRRA GDF6 HOXA6 IL17F ADNP BRPF3 CERS1 CX3CL1 ETS1 GIN1 HOXA7 IL18 AEBP1 BUD31 CERS2 CXCL10 ETS2 GLIS3 HOXB1 IL19 AFF4 C17ORF77 CERS4 CXCL11 ETV3 GMEB1 HOXB13 IL1A AHR C1QTNF4 CFL2 CXCL12 ETV7 GPBP1 HOXB5 IL1B AIMP1 C21ORF66 CHIA CXCL13 FAM3B GPER HOXB6 IL1F3 ALS2CR8 CBFA2T2 CIR1 CXCL14 FAM3D GPI HOXB7 IL1F5 ALX1 CBFA2T3 CITED1 CXCL16 FASLG GREM1 HOXB9 IL1F6 ARGFX CBFB CITED2 CXCL3 FBLN1 GREM2 HOXC4 IL1F7 -

Genome-Wide DNA Methylation Analysis of KRAS Mutant Cell Lines Ben Yi Tew1,5, Joel K
www.nature.com/scientificreports OPEN Genome-wide DNA methylation analysis of KRAS mutant cell lines Ben Yi Tew1,5, Joel K. Durand2,5, Kirsten L. Bryant2, Tikvah K. Hayes2, Sen Peng3, Nhan L. Tran4, Gerald C. Gooden1, David N. Buckley1, Channing J. Der2, Albert S. Baldwin2 ✉ & Bodour Salhia1 ✉ Oncogenic RAS mutations are associated with DNA methylation changes that alter gene expression to drive cancer. Recent studies suggest that DNA methylation changes may be stochastic in nature, while other groups propose distinct signaling pathways responsible for aberrant methylation. Better understanding of DNA methylation events associated with oncogenic KRAS expression could enhance therapeutic approaches. Here we analyzed the basal CpG methylation of 11 KRAS-mutant and dependent pancreatic cancer cell lines and observed strikingly similar methylation patterns. KRAS knockdown resulted in unique methylation changes with limited overlap between each cell line. In KRAS-mutant Pa16C pancreatic cancer cells, while KRAS knockdown resulted in over 8,000 diferentially methylated (DM) CpGs, treatment with the ERK1/2-selective inhibitor SCH772984 showed less than 40 DM CpGs, suggesting that ERK is not a broadly active driver of KRAS-associated DNA methylation. KRAS G12V overexpression in an isogenic lung model reveals >50,600 DM CpGs compared to non-transformed controls. In lung and pancreatic cells, gene ontology analyses of DM promoters show an enrichment for genes involved in diferentiation and development. Taken all together, KRAS-mediated DNA methylation are stochastic and independent of canonical downstream efector signaling. These epigenetically altered genes associated with KRAS expression could represent potential therapeutic targets in KRAS-driven cancer. Activating KRAS mutations can be found in nearly 25 percent of all cancers1. -

Alveolar Rhabdomyosarcoma-Associated Proteins PAX3/FOXO1A and PAX7/FOXO1A Suppress the Transcriptional Activity of Myod-Target Genes in Muscle Stem Cells
Oncogene (2013) 32, 651 --662 & 2013 Macmillan Publishers Limited All rights reserved 0950-9232/13 www.nature.com/onc ORIGINAL ARTICLE Alveolar rhabdomyosarcoma-associated proteins PAX3/FOXO1A and PAX7/FOXO1A suppress the transcriptional activity of MyoD-target genes in muscle stem cells F Calhabeu1, S Hayashi2, JE Morgan3, F Relaix2 and PS Zammit1 Rhabdomyosarcoma (RMS) is the commonest soft-tissue sarcoma in childhood and is characterized by expression of myogenic proteins, including the transcription factors MyoD and myogenin. There are two main subgroups, embryonal RMS and alveolar RMS (ARMS). Most ARMS are associated with chromosomal translocations that have breakpoints in introns of either PAX3 or PAX7, and FOXO1A. These translocations create chimeric transcription factors termed PAX3/FOXO1A and PAX7/FOXO1A respectively. Upon ectopic PAX3/FOXO1A expression, together with other genetic manipulation in mice, both differentiating myoblasts and satellite cells (the resident stem cells of postnatal muscle) can give rise to tumours with ARMS characteristics. As PAX3 and PAX7 are part of transcriptional networks that regulate muscle stem cell function in utero and during early postnatal life, PAX3/FOXO1A and PAX7/FOXO1A may subvert normal PAX3 and PAX7 functions. Here we examined how PAX3/FOXO1A and PAX7/FOXO1A affect myogenesis in satellite cells. PAX3/FOXO1A or PAX7/FOXO1A inhibited myogenin expression and prevented terminal differentiation in murine satellite cells: the same effect as dominant-negative (DN) Pax3 or Pax7 constructs. The transcription of MyoD-target genes myogenin and muscle creatine kinase were suppressed by PAX3/FOXO1A or PAX7/ FOXO1A in C2C12 myogenic cells again as seen with Pax3/7DN. PAX3/FOXO1A or PAX7/FOXO1A did not inhibit the transcriptional activity of MyoD by perturbing MyoD expression, localization, phosphorylation or interaction with E-proteins. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Transcriptomic and Proteomic Profiling Provides Insight Into
BASIC RESEARCH www.jasn.org Transcriptomic and Proteomic Profiling Provides Insight into Mesangial Cell Function in IgA Nephropathy † † ‡ Peidi Liu,* Emelie Lassén,* Viji Nair, Celine C. Berthier, Miyuki Suguro, Carina Sihlbom,§ † | † Matthias Kretzler, Christer Betsholtz, ¶ Börje Haraldsson,* Wenjun Ju, Kerstin Ebefors,* and Jenny Nyström* *Department of Physiology, Institute of Neuroscience and Physiology, §Proteomics Core Facility at University of Gothenburg, University of Gothenburg, Gothenburg, Sweden; †Division of Nephrology, Department of Internal Medicine and Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, Michigan; ‡Division of Molecular Medicine, Aichi Cancer Center Research Institute, Nagoya, Japan; |Department of Immunology, Genetics and Pathology, Uppsala University, Uppsala, Sweden; and ¶Integrated Cardio Metabolic Centre, Karolinska Institutet Novum, Huddinge, Sweden ABSTRACT IgA nephropathy (IgAN), the most common GN worldwide, is characterized by circulating galactose-deficient IgA (gd-IgA) that forms immune complexes. The immune complexes are deposited in the glomerular mesangium, leading to inflammation and loss of renal function, but the complete pathophysiology of the disease is not understood. Using an integrated global transcriptomic and proteomic profiling approach, we investigated the role of the mesangium in the onset and progression of IgAN. Global gene expression was investigated by microarray analysis of the glomerular compartment of renal biopsy specimens from patients with IgAN (n=19) and controls (n=22). Using curated glomerular cell type–specific genes from the published literature, we found differential expression of a much higher percentage of mesangial cell–positive standard genes than podocyte-positive standard genes in IgAN. Principal coordinate analysis of expression data revealed clear separation of patient and control samples on the basis of mesangial but not podocyte cell–positive standard genes. -

Pig Antibodies
Pig Antibodies gene_name sku Entry_Name Protein_Names Organism Length Identity CDX‐2 ARP31476_P050 D0V4H7_PIG Caudal type homeobox 2 (Fragment) Sus scrofa (Pig) 147 100.00% CDX‐2 ARP31476_P050 A7MAE3_PIG Caudal type homeobox transcription factor 2 (Fragment) Sus scrofa (Pig) 75 100.00% Tnnt3 ARP51286_P050 Q75NH3_PIG Troponin T fast skeletal muscle type Sus scrofa (Pig) 271 85.00% Tnnt3 ARP51286_P050 Q75NH2_PIG Troponin T fast skeletal muscle type Sus scrofa (Pig) 266 85.00% Tnnt3 ARP51286_P050 Q75NH1_PIG Troponin T fast skeletal muscle type Sus scrofa (Pig) 260 85.00% Tnnt3 ARP51286_P050 Q75NH0_PIG Troponin T fast skeletal muscle type Sus scrofa (Pig) 250 85.00% Tnnt3 ARP51286_P050 Q75NG8_PIG Troponin T fast skeletal muscle type Sus scrofa (Pig) 266 85.00% Tnnt3 ARP51286_P050 Q75NG7_PIG Troponin T fast skeletal muscle type Sus scrofa (Pig) 260 85.00% Tnnt3 ARP51286_P050 Q75NG6_PIG Troponin T fast skeletal muscle type Sus scrofa (Pig) 250 85.00% Tnnt3 ARP51286_P050 TNNT3_PIG Troponin T, fast skeletal muscle (TnTf) Sus scrofa (Pig) 271 85.00% ORF Names:PANDA_000462 EMBL EFB13877.1OrganismAiluropod High mobility group protein B2 (High mobility group protein a melanoleuca (Giant panda) ARP31939_P050 HMGB2_PIG 2) (HMG‐2) Sus scrofa (Pig) 210 100.00% Agpat5 ARP47429_P050 B8XTR3_PIG 1‐acylglycerol‐3‐phosphate O‐acyltransferase 5 Sus scrofa (Pig) 365 85.00% irf9 ARP31200_P050 Q29390_PIG Transcriptional regulator ISGF3 gamma subunit (Fragment) Sus scrofa (Pig) 57 100.00% irf9 ARP31200_P050 Q0GFA1_PIG Interferon regulatory factor 9 Sus scrofa (Pig) -

(12) Patent Application Publication (10) Pub. No.: US 2011/001412.6 A1 Evans Et Al
US 2011 0014126A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2011/001412.6 A1 Evans et al. (43) Pub. Date: Jan. 20, 2011 (54) USE OF VITAMIND RECEPTORAGONISTS (60) Provisional application No. 60/985,972, filed on Nov. AND PRECURSORS TO TREAT FIBROSS 6, 2007. (76) Inventors: Ronald M. Evans, La Jolla, CA Publication Classification (US); Michael Downes, San Diego, CA (US); Christopher Liddle, (51) Int. Cl. New South Wales (AU): A 6LX 3/59 (2006.01) Nanthakumar Subramaniam, A6IPL/I6 (2006.01) New South Wales (AU); Caroline CI2O 1/02 (2006.01) Flora Samer, Geneva (CH) A61R 49/00 (2006.01) CI2N 5/071 (2010.01) Correspondence Address: (52) U.S. Cl. ............. 424/9.2: 514/167; 435/29: 435/375 KLARQUIST SPARKMAN, LLP 121 S.W. SALMONSTREET, SUITE 1600 (57) ABSTRACT PORTLAND, OR 97204 (US) This application relates to methods of treating, preventing, (21) Appl. No.: 12/772,981 and ameliorating fibrosis, such as fibrosis of the liver. In particular, the application relates to methods of using a vita (22) Filed: May 3, 2010 min D receptor agonist (Such as vitamin D. Vitamin Dana logs, vitamin D precursors, and vitamin D receptor agonists Related U.S. Application Data precursors) for the treatment of liver fibrosis. Also disclosed (63) Continuation-in-part of application No. 12/266,513, are methods for screening for agents that treat, prevent, and filed on Nov. 6, 2008. ameliorate fibrosis. Stellate Cells Liver RR1,3 AR, ERa ERR1,2,3, AR, ERa CNF GR, MR CNF RARa, GR, MR NF4g Rab NF4ag RARa,b, NOR1 a, RH1 TRa,b WDR NURR1 NOR1 RORa,b,g RORag CAR Receptor SF-1 FXRa,b FXRa,b epissertoup. -

Transcriptional Code and Disease Map for Adult Retinal Cell Types
RE so UR C E Transcriptional code and disease map for adult retinal cell types Sandra Siegert1,7, Erik Cabuy1,7, Brigitte Gross Scherf1, Hubertus Kohler1, Satchidananda Panda2, Yun-Zheng Le3,4, Hans Jörg Fehling5, Dimos Gaidatzis1,6, Michael B Stadler1,6 & Botond Roska1 Brain circuits are assembled from a large variety of morphologically and functionally diverse cell types. It is not known how the intermingled cell types of an individual adult brain region differ in their expressed genomes. Here we describe an atlas of cell type transcriptomes in one brain region, the mouse retina. We found that each adult cell type expressed a specific set of genes, including a unique set of transcription factors, forming a ‘barcode’ for cell identity. Cell type transcriptomes carried enough information to categorize cells into morphological classes and types. Several genes that were specifically expressed in particular retinal circuit elements, such as inhibitory neuron types, are associated with eye diseases. The resource described here allows gene expression to be compared across adult retinal cell types, experimenting with specific transcription factors to differentiate stem or somatic cells to retinal cell types, and predicting cellular targets of newly discovered disease-associated genes. The brain is composed of many neuronal cell types that are deter- across the retina with a mosaic-like distribution (Supplementary Text mined during development by a dynamic transcriptional program1–5. and Supplementary Fig. 1). The cellular architecture of the retina is In adults, neurons sampled from different brain areas such as the highly conserved among mammals14–17. cortex, cerebellum and hippocampus maintain differences in their By constructing and analyzing a transcriptome atlas for retinal cell expressed genomes6,7. -

Engineered Type 1 Regulatory T Cells Designed for Clinical Use Kill Primary
ARTICLE Acute Myeloid Leukemia Engineered type 1 regulatory T cells designed Ferrata Storti Foundation for clinical use kill primary pediatric acute myeloid leukemia cells Brandon Cieniewicz,1* Molly Javier Uyeda,1,2* Ping (Pauline) Chen,1 Ece Canan Sayitoglu,1 Jeffrey Mao-Hwa Liu,1 Grazia Andolfi,3 Katharine Greenthal,1 Alice Bertaina,1,4 Silvia Gregori,3 Rosa Bacchetta,1,4 Norman James Lacayo,1 Alma-Martina Cepika1,4# and Maria Grazia Roncarolo1,2,4# Haematologica 2021 Volume 106(10):2588-2597 1Department of Pediatrics, Division of Stem Cell Transplantation and Regenerative Medicine, Stanford School of Medicine, Stanford, CA, USA; 2Stanford Institute for Stem Cell Biology and Regenerative Medicine, Stanford School of Medicine, Stanford, CA, USA; 3San Raffaele Telethon Institute for Gene Therapy, Milan, Italy and 4Center for Definitive and Curative Medicine, Stanford School of Medicine, Stanford, CA, USA *BC and MJU contributed equally as co-first authors #AMC and MGR contributed equally as co-senior authors ABSTRACT ype 1 regulatory (Tr1) T cells induced by enforced expression of interleukin-10 (LV-10) are being developed as a novel treatment for Tchemotherapy-resistant myeloid leukemias. In vivo, LV-10 cells do not cause graft-versus-host disease while mediating graft-versus-leukemia effect against adult acute myeloid leukemia (AML). Since pediatric AML (pAML) and adult AML are different on a genetic and epigenetic level, we investigate herein whether LV-10 cells also efficiently kill pAML cells. We show that the majority of primary pAML are killed by LV-10 cells, with different levels of sensitivity to killing. Transcriptionally, pAML sensitive to LV-10 killing expressed a myeloid maturation signature. -
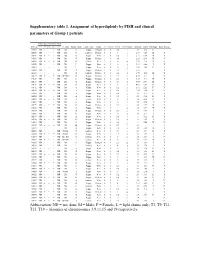
Supplementary Table 1. Assignment of Hyperdiploidy by FISH and Clinical Parameters of Group 1 Patients
Supplementary table 1. Assignment of hyperdiploidy by FISH and clinical parameters of Group 1 patients FISH Based Trisomy Index MAYO T3 T9 T11 T15 T19 TC class Heavy Chain Light Chain Status 13 VH/CH PCLI GEP Cluster Albumin B2M ISS Stage Bone Disease M003 ND + + + ND D1 G Kappa Relapse 0 0 4.2 1 3.4 4.16 II - M029 ND + - + ND D1 G lambda Relapse 0 0 3 1 2.74 7.29 III Y M037 ND + + - ND D2 G Kappa New 1 0 0.2 2 2.77 6.54 III Y M051 ND + - + ND D2 G Kappa Relapse 1 0 4.8 1 3.24 8.46 III Y M058 ND + + + ND D1 G lambda New 1 0 0 4 3.15 7.18 III N M059 ND + - + ND D1 L Kappa New 0 1 0 4 3.94 2.28 I Y M061 + + - - - D1 L Kappa New 0 0 0 2 2.67 2.61 II Y M082 ND + + - ND D2 A Kappa Relapse 1 0 5.8 1 2.76 - - Y M113 + + - - - D1 G lambda Relapse 0 0 2.2 4 3.79 11.6 III Y M123 ND + - + ND D1+D2 G Kappa Relapse 1 1 1.2 2 4.28 1.7 I Y M128 ND + + + ND D1 L Kappa Relapse 0 1 2.3 4 4.17 7.4 III Y M131 ND - + + ND D1 G Kappa Relapse 0 0 4.4 4 4.88 8.74 III Y M135 ND + + + ND D1 G Kappa New 0 0 0 3 4.01 5.23 II Y M142 ND + + - ND D1 A Kappa New 0 0 0.2 3 3.92 5.23 II Y M163 ND + + + ND D1 A Kappa New 0 0 0.4 3 2.96 2.06 II N M198 ND + + + ND D1 A Kappa New 0 0 0.4 2 3.1 3.3 II Y M200 ND + + - ND D2 A Kappa New 0 0 3.8 1 2.5 3.16 II Y M218 ND + + + ND D1 A lambda New 0 0 0 3 3.6 4.31 II Y M227 ND - + + ND D1 L Kappa New 1 0 0 3 3.9 2.76 I Y M229 ND - + + ND D1 G Kappa Relapse 0 0 3.8 1 4.3 8.77 III Y M230 ND + + - ND D1 A Kappa New 1 1 1 4 3.6 6.94 III Y M235 ND + + + ND D1 G Kappa Relapse 0 0 0.2 2 4 2.37 I Y M245 ND + - + ND D1 G Kappa New 0 0 0 4