Transcriptome Analysis and Metabolite Profiling for Dissection Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Electronic Supplementary Material (ESI) for Molecular Biosystems
Electronic Supplementary Material (ESI) for Molecular BioSystems. This journal is © The Royal Society of Chemistry 2015 Table S5 Mass data of proteins identified with label free shotgun proteomics a #Alt. Proteins; b Scores; c #Peptides; d SC [%]; e RMS90 [ppm]; f Rank; g Median(Controls:GLP1); h #(Controls:GLP1); i CV [%](Controls:GLP1); l Median(Controls:Palmitate); m #(Controls:Palmitate) ; n CV [%](Controls:Palmitate); o Median(Controls:GLP1 + Palmitate); p #(Controls:GLP1 + Palmitate); q CV [%](Controls:GLP1 + Palmitate) MW OK Accession Protein pI a b c d e f g h i l m n o p q [kDa] 78 kDa glucose-regulated protein OS=Rattus 1672.6 true GRP78_RAT 72.3 4.9 1 22 39 1.48 1 1.16 6 20.53 1.18 9 9.56 1.24 10 13.52 norvegicus GN=Hspa5 PE=1 SV=1 (M:1672.6) Endoplasmin OS=Rattus norvegicus 1253.0 true ENPL_RAT 92.7 4.6 1 23 29 3.34 2 0.85 8 33.34 1.19 6 39.58 1.18 10 28.11 GN=Hsp90b1 PE=1 SV=2 (M:1253.0) Stress-70 protein, mitochondrial OS=Rattus 1138.8 true GRP75_RAT 73.8 5.9 1 17 28.7 1.18 3 1.21 5 7.62 0.78 1 1.1 8 8.04 norvegicus GN=Hspa9 PE=1 SV=3 (M:1138.8) Protein disulfide-isomerase A3 OS=Rattus 1035.4 true PDIA3_RAT 56.6 5.8 1 16 35.2 2.84 4 0.94 3 17.22 1.15 4 26.62 1.25 4 7.86 norvegicus GN=Pdia3 PE=1 SV=2 (M:1035.4) Aconitate hydratase, mitochondrial 983.8 true ACON_RAT OS=Rattus norvegicus GN=Aco2 PE=1 85.4 8.7 1 19 31.4 2.72 5 0.89 2 23.6 1.21 2 9.25 0.89 3 32.87 (M:983.8) SV=2 60 kDa heat shock protein, mitochondrial 906.9 true CH60_RAT OS=Rattus norvegicus GN=Hspd1 PE=1 60.9 5.8 1 13 32.5 3.26 6 1.05 5 13.15 1.01 2 32.54 1.01 -

Murine Neonatal Ketogenesis Preserves Mitochondrial Energetics by Preventing Protein Hyperacetylation
ARTICLES https://doi.org/10.1038/s42255-021-00342-6 Murine neonatal ketogenesis preserves mitochondrial energetics by preventing protein hyperacetylation Yuichiro Arima 1,2,13 ✉ , Yoshiko Nakagawa3,13, Toru Takeo 3,13, Toshifumi Ishida 1, Toshihiro Yamada1, Shinjiro Hino4, Mitsuyoshi Nakao4, Sanshiro Hanada 2, Terumasa Umemoto 2, Toshio Suda2, Tetsushi Sakuma 5, Takashi Yamamoto5, Takehisa Watanabe6, Katsuya Nagaoka6, Yasuhito Tanaka6, Yumiko K. Kawamura7,8, Kazuo Tonami7, Hiroki Kurihara7, Yoshifumi Sato9, Kazuya Yamagata9,10, Taishi Nakamura 1,11, Satoshi Araki1, Eiichiro Yamamoto1, Yasuhiro Izumiya1,12, Kenji Sakamoto1, Koichi Kaikita1, Kenichi Matsushita 1, Koichi Nishiyama2, Naomi Nakagata3 and Kenichi Tsujita1,10 Ketone bodies are generated in the liver and allow for the maintenance of systemic caloric and energy homeostasis during fasting and caloric restriction. It has previously been demonstrated that neonatal ketogenesis is activated independently of starvation. However, the role of ketogenesis during the perinatal period remains unclear. Here, we show that neonatal ketogen- esis plays a protective role in mitochondrial function. We generated a mouse model of insufficient ketogenesis by disrupting the rate-limiting hydroxymethylglutaryl-CoA synthase 2 enzyme gene (Hmgcs2). Hmgcs2 knockout (KO) neonates develop microvesicular steatosis within a few days of birth. Electron microscopic analysis and metabolite profiling indicate a restricted energy production capacity and accumulation of acetyl-CoA in Hmgcs2 KO mice. Furthermore, -
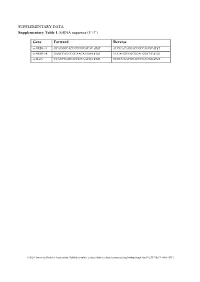
Supplementary Tables and Figures
SUPPLEMENTARY DATA Supplementary Table 1. SiRNA sequence (5’-3’) Gene Forward Reverse si-HRD1-1# GCAUGGCAGUCCUGUACAU dTdT AUGUACAGGACUGCCAUGC dTdT si-HRD1-2# GAGCCAUCCGCAACAUGAA dTdT UUCAUGUUGCGGAUGGCUC dTdT si-MafA CCAUCGAGUACGUCAACGA dTdT UCGUUGACGUACUCGAUGG dTdT ©2020 American Diabetes Association. Published online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db19-1060/-/DC1 SUPPLEMENTARY DATA Supplementary Table 2. Primer sequences for qRT-PCR (5’-3’) Gene Forward Reverse human HRD1 GCTCACGCCTACTACCTCAAA GCCAGACAAGTCTCTGTGACG mouse mafA AAGCGGCGCACGCTCAAGAA GGTCCCGCTCCTTGGCCAGA mouse insulin1 CACTTCCTACCCCTGCTGG ACCACAAAGATGCTGTTTGACA mouse β-actin AGGCCAACCGTGAAAAGATG AGAGCATAGCCCTCGTAGATGG human β-actin CATGTACGTTGCTATCCAGGC CTCCTTAATGTCACGCACGAT ©2020 American Diabetes Association. Published online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db19-1060/-/DC1 SUPPLEMENTARY DATA Supplementary Table 3. Primer sequences for ChIP (5’-3’) Gene promoter Forward Reverse mouse Insulin1, 2 GGAACTGTGAAACAGTCCAAGG CCCCCTGGACTTTGCTGTTTG ©2020 American Diabetes Association. Published online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db19-1060/-/DC1 SUPPLEMENTARY DATA Supplementary Table 4. Primer sequences for PCR (5’-3’) Gene Forward Reverse HRD1-pDsred CCCAAGCTTATGTTCCGCACCGCAGT GGGGTACCCAGTGGGCAACAGGGG HRD1-pCMV- Flag GGGGTACCATGTTCCGCACCGCAGT CCCAAGCTTGTGGGCAACAGGGGACT C HRD1-pCMV-HA GGCCATGGGCCATATGGGATCCTTCC AGGGATGCCACCCGGGGATCCTCAGT GCACCGCAGTGATG GGGCAACAGGGGAC HRD1-N-HA GGCCATGGGCCATATGGGATCCTTCC -

KENNETH B. STOREY and ERNEST BAILEY Department of Biochemistry, University of Sheffield, Sheffield SI0 2TN, U.K
Insect Biochem.,. 1978, Vol. 8, pp. 125 to 131. Peroamon Press. Printed in Great Britain INTRACELLULAR DISTRIBUTION OF ENZYMES ASSOCIATED WITH LIPOGENESIS AND GLUCONEOGENESIS IN FAT BODY OF THE ADULT COCKROACH, PERIPLANETA KENNETH B. STOREY and ERNEST BAILEY Department of Biochemistry, University of Sheffield, Sheffield SI0 2TN, U.K. (Received 30 April 1977) Abstract--The intracellular distribution and maximal activities of some enzymes associated with lipo- genesis, gluconeogenesis and fatty acid oxidation have been determined in the fat body of the adult male cockroach Periplaneta americana. Of the enzymes of lipogenesis, acetyl-CoA synthase, acetyl-CoA carboxylase, glucose 6-phosphate dehydrogenase, and ATP citrate lyase are located entirely in the cytosol. Of the other enzymes of citrate metabolism studied, citrate synthase and NAD-dependent isocitrate dehydrogenase are almost exclusively mitochondrial, whereas NADP-dependent isocitrate dehydrogenase and aconitase are predominantly cytosolic although significant mitochondrial activity is also present. The latter subeellular distribution is also observed for 'malic enzyme' and NAD and NADP dependent malate dehydrogenase. The enzyme of fatty acid oxidation studied, 3-hydroxyacyl- CoA dehydrogenase is entirely mitochondrial. Of the enzymes possibly involved in glueoneogenesis, glucose 6-phosphate is microsomal, fructose 1,6-diphosphatase cytosolic, pyruvate carboxylase mito- chondrial and phosphoenolpyruvate carboxykinase predominantly eytosolic. Of the enzymes of amino acid metabolism studied, glutamate dehydrogenase is NAD-dependent and located in the mitochondria whereas glutamate/oxalacetate and glutamate/pyruvate transaminases are predominantly cytosolic but with significant activity in the mitochondria. Glycerol kinase and sorbitol dehydrogenase are cytosolic and the glyoxylate cycle enzymes malate synthetase and isocitrate lyase are not detected in the fat body. -

Candidate Genes Linked to QTL Regions Associated with Fatty Acid
1 Candidate genes linked to QTL regions associated with fatty acid composition in oil palm 2 3 Abstract 4 5 The present study searched for candidate genes in five linkage groups (LGs) - T2, T3, OT4, OT6 and T9 hosting the QTLs 6 associated with iodine value (IV) and fatty acid composition (FAC) in an oil palm interspecific hybrid population. Each 7 of the five LGs was successfully anchored to its corresponding chromosomal segment where, a wider repertoire of 8 candidate genes was identified. This study further revealed a total of 19 candidate genes and four transcription factors 9 involved in biosynthesis of fatty acids, lipids (including triacylglycerol) and acetyl-CoA, glycosylation and degradation 10 of fatty acids. Their possible involvement in regulating the levels of saturation are discussed. In addition, 22 candidate 11 genes located outside the QTL intervals were also identified across the interspecific hybrid genome. A total of 92 SSR 12 markers were developed to tag the presence of these candidate genes and 50 were successfully mapped onto their 13 respective positions on the genome. The data obtained here complements the previous studies, and collectively, these 14 QTL-linked candidate gene markers could help breeders in more precisely selecting palms with the desired FAC. 15 16 Keywords 17 18 Elaeis guineensis, interspecific hybrid, fatty acid biosynthesis, triacylglycerol biosynthesis, transcription factors, genetic 19 linkage map 20 21 Abbreviations 22 23 4CLL1 4-coumarate--CoA ligase 1 24 AACT acetoacetyl-CoA thiolase 25 acbd4 acyl-CoA-binding domain-containing protein 4 26 ACX4 acyl-CoA oxidase 4 27 C14:0 myristic acid 28 C16:0 palmitic acid 29 C16:1 palmitoleic acid 30 C18:0 stearic acid 31 C18:1 oleic acid 32 C18:2 linoleic acid 33 CHR pseudo-chromosome 34 cM centiMorgan 35 CP cross pollinator 36 CPO crude palm oil 37 CTAB cetyltriammonium bromide 38 DNA deoxyribonucleic acid 39 EG5 E. -

FABP7 Regulates Acetyl-Coa Metabolism Through the Interaction with ACLY in the Nucleus of Astrocytes
Molecular Neurobiology (2020) 57:4891–4910 https://doi.org/10.1007/s12035-020-02057-3 FABP7 Regulates Acetyl-CoA Metabolism Through the Interaction with ACLY in the Nucleus of Astrocytes Yoshiteru Kagawa1 & Banlanjo Abdulaziz Umaru1 & Hiroki Shima2 & Ryo Ito3 & Ryo Zama1 & Ariful Islam1 & Shin-ichiro Kanno4 & Akira Yasui4 & Shun Sato5 & Kosuke Jozaki5 & Subrata Kumar Shil1 & Hirofumi Miyazaki1 & Shuhei Kobayashi1 & Yui Yamamoto1 & Hiroshi Kogo6 & Chie Shimamoto-Mitsuyama7 & Akira Sugawara3 & Norihiro Sugino5 & Masayuki Kanamori8 & Teiji Tominaga8 & Takeo Yoshikawa7 & Kohji Fukunaga9 & Kazuhiko Igarashi2 & Yuji Owada1 Received: 27 April 2020 /Accepted: 7 August 2020 / Published online: 19 August 2020 # The Author(s) 2020 Abstract Fatty acid binding protein 7 (FABP7) is an intracellular fatty acid chaperon that is highly expressed in astrocytes, oligodendrocyte-precursor cells, and malignant glioma. Previously, we reported that FABP7 regulates the response to extracel- lular stimuli by controlling the expression of caveolin-1, an important component of lipid raft. Here, we explored the detailed mechanisms underlying FABP7 regulation of caveolin-1 expression using primary cultured FABP7-KO astrocytes as a model of loss of function and NIH-3T3 cells as a model of gain of function. We discovered that FABP7 interacts with ATP-citrate lyase (ACLY) and is important for acetyl-CoA metabolism in the nucleus. This interaction leads to epigenetic regulation of several genes, including caveolin-1. Our novel findings suggest that FABP7-ACLY modulation of nuclear acetyl-CoA has more influence on histone acetylation than cytoplasmic acetyl-CoA. The changes to histone structure may modify caveolae-related cell activity in astrocytes and tumors, including malignant glioma. Keywords Fatty acid–binding protein (FABP) . -

Wo 2011/063350 A2
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date 26 May 2011 (26.05.2011) WO 2011/063350 A2 (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every CI2N 1/00 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, (21) Number: International Application CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, PCT/US2010/057668 DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, 22 November 2010 (22.1 1.2010) KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, (25) Filing Language: English NO, NZ, OM, PE, PG, PH, PL, PT, RO, RS, RU, SC, SD, (26) Publication Language: English SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: 61/263,775 23 November 2009 (23.1 1.2009) US (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (71) Applicant (for all designated States except US): CIBUS GM, KE, LR, LS, MW, MZ, NA, SD, SL, SZ, TZ, UG, OILS, LLC [US/US]; 6455 Nancy Ridge Drive, Suite ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, RU, TJ, 100, San Diego, California 92121 (US). -

Kidney V-Atpase-Rich Cell Proteome Database
A comprehensive list of the proteins that are expressed in V-ATPase-rich cells harvested from the kidneys based on the isolation by enzymatic digestion and fluorescence-activated cell sorting (FACS) from transgenic B1-EGFP mice, which express EGFP under the control of the promoter of the V-ATPase-B1 subunit. In these mice, type A and B intercalated cells and connecting segment principal cells of the kidney express EGFP. The protein identification was performed by LC-MS/MS using an LTQ tandem mass spectrometer (Thermo Fisher Scientific). For questions or comments please contact Sylvie Breton ([email protected]) or Mark A. Knepper ([email protected]). -

Suberin Biosynthesis and Deposition in the Wound-Healing Potato (Solanum Tuberosum L.) Tuber Model
Western University Scholarship@Western Electronic Thesis and Dissertation Repository 12-4-2018 2:30 PM Suberin Biosynthesis and Deposition in the Wound-Healing Potato (Solanum tuberosum L.) Tuber Model Kathlyn Natalie Woolfson The University of Western Ontario Supervisor Bernards, Mark A. The University of Western Ontario Graduate Program in Biology A thesis submitted in partial fulfillment of the equirr ements for the degree in Doctor of Philosophy © Kathlyn Natalie Woolfson 2018 Follow this and additional works at: https://ir.lib.uwo.ca/etd Part of the Plant Biology Commons Recommended Citation Woolfson, Kathlyn Natalie, "Suberin Biosynthesis and Deposition in the Wound-Healing Potato (Solanum tuberosum L.) Tuber Model" (2018). Electronic Thesis and Dissertation Repository. 5935. https://ir.lib.uwo.ca/etd/5935 This Dissertation/Thesis is brought to you for free and open access by Scholarship@Western. It has been accepted for inclusion in Electronic Thesis and Dissertation Repository by an authorized administrator of Scholarship@Western. For more information, please contact [email protected]. Abstract Suberin is a heteropolymer comprising a cell wall-bound poly(phenolic) domain (SPPD) covalently linked to a poly(aliphatic) domain (SPAD) that is deposited between the cell wall and plasma membrane. Potato tuber skin contains suberin to protect against water loss and microbial infection. Wounding triggers suberin biosynthesis in usually non- suberized tuber parenchyma, providing a model system to study suberin production. Spatial and temporal coordination of SPPD and SPAD-related metabolism are required for suberization, as the former is produced soon after wounding, and the latter is synthesized later into wound-healing. Many steps involved in suberin biosynthesis remain uncharacterized, and the mechanism(s) that regulate and coordinate SPPD and SPAD production and assembly are not understood. -

Metabolic Investigations of the Molecular Mechanisms Associated with Parkinson’S Disease
H OH metabolites OH Review Metabolic Investigations of the Molecular Mechanisms Associated with Parkinson’s Disease Robert Powers 1,2,*, Shulei Lei 1, Annadurai Anandhan 3,4, Darrell D. Marshall 1, Bradley Worley 1, Ronald L. Cerny 1, Eric D. Dodds 1, Yuting Huang 1, Mihalis I. Panayiotidis 5, Aglaia Pappa 6 and Rodrigo Franco 3,4,* 1 Department of Chemistry, University of Nebraska-Lincoln, Lincoln, NE 68588, USA; [email protected] (S.L.); [email protected] (D.D.M.); [email protected] (B.W.); [email protected] (R.L.C.); [email protected] (E.D.D.); [email protected] (Y.H.) 2 Department of Biochemistry, University of Nebraska-Lincoln, Lincoln, NE 68588, USA 3 Redox Biology Center, University of Nebraska-Lincoln, Lincoln, NE 68588, USA; [email protected] 4 School of Veterinary Medicine and Biomedical Sciences, University of Nebraska-Lincoln, Lincoln, NE 68588, USA 5 Department of Applied Sciences, Northumbria University, Newcastle Upon Tyne NE1 8ST, UK; [email protected] 6 Department of Molecular Biology and Genetics, Democritus University of Thrace, Alexandroupolis 68100, Greece; [email protected] * Correspondence: [email protected] (R.P.); [email protected] (R.F.); Tel.: +01-402-472-3039 (R.P.); +01-402-472-8547 (R.F.) Academic Editors: Daniel Raftery, James Cox, Katja Dettmer and Adrian S. Culf Received: 5 April 2017; Accepted: 16 May 2017; Published: 24 May 2017 Abstract: Parkinson’s disease (PD) is a neurodegenerative disorder characterized by fibrillar cytoplasmic aggregates of α-synuclein (i.e., Lewy bodies) and the associated loss of dopaminergic cells in the substantia nigra. -

ACLY Antibody
Efficient Professional Protein and Antibody Platforms ACLY Antibody Basic information: Catalog No.: UPA62638 Source: Rabbit Size: 50ul/100ul Clonality: Monoclonal Concentration: 1mg/ml Isotype: Rabbit IgG Purification: Protein A affinity purified Useful Information: WB:1:1000-1:2000 ICC:1:50-1:200 Applications: IHC:1:50-1:200 FC:1:50-1:100 Reactivity: Human, Mouse, Rat Specificity: This antibody recognizes ACLY protein. Immunogen: recombinant protein ATP-citrate synthase, also designated ATP-citrate lyase or citrate cleavage enzyme, is a cytoplasmic homotetramer belonging to the succinate/malate CoA ligase family. The gene coding for this protein maps against chromo- Description: some 17q12-q21. ATP-citrate synthase catalyses the formation of acetyl-CoA and oxaloacetate from citrate and CoA. This product, Acetyl-CoA, is neces- sary for both fatty acid and cholesterol biosynthesis. ATP citrate-lyase is im- portant in the biosynthesis of acetylcholine in nervous tissue. Uniprot: P53396(Human) Q91V92(Mouse) P16638(Rat) BiowMW: 122 kDa Buffer: 1*TBS (pH7.4), 1%BSA, 40%Glycerol. Preservative: 0.05% Sodium Azide. Storage: Store at 4°C short term and -20°C long term. Avoid freeze-thaw cycles. Note: For research use only, not for use in diagnostic procedure. Data: Western blot analysis of ATP citrate lyase on dif- ferent lysates using anti-ATP citrate lyase anti- body at 1/1,000 dilution. Positive control: Lane 1: Mouse kidney Lane 2: Mouse colon Lane 3: MCF-7 Gene Universal Technology Co. Ltd www.universalbiol.com Tel: 0550-3121009 E-mail: [email protected] Efficient Professional Protein and Antibody Platforms ICC staining ATP citrate lyase in Hela cells (green). -

Linking Metabolic Reprogramming, Plasticity and Tumor Progression
cancers Review Linking Metabolic Reprogramming, Plasticity and Tumor Progression Oleg Shuvalov 1 , Alexandra Daks 1, Olga Fedorova 1, Alexey Petukhov 1,2 and Nickolai Barlev 1,3,4,* 1 Institute of Cytology RAS, 194064 St-Petersburg, Russia; [email protected] (O.S.); [email protected] (A.D.); [email protected] (O.F.); [email protected] (A.P.) 2 Almazov National Medical Research Center, 197341 St-Petersburg, Russia 3 MIPT, 141701 Dolgoprudny, Moscow Region, Russia 4 Orekhovich IBMC, 119435 Moscow, Russia * Correspondence: [email protected]; Tel.: +7-812-297-4519 Simple Summary: In the present review, we discuss the role of metabolic reprogramming which occurs in malignant cells. The process of metabolic reprogramming is also known as one of the “hallmarks of cancer”. Due to several reasons, including the origin of cancer, tumor microenvi- ronment, and the tumor progression stage, metabolic reprogramming can be heterogeneous and dynamic. In this review, we provide evidence that the usage of metabolic drugs is a promising approach to treat cancer. However, because these drugs can damage not only malignant cells but also normal rapidly dividing cells, it is important to understand the exact metabolic changes which are elicited by particular drivers in concrete tissue and are specific for each stage of cancer development, including metastases. Finally, the review highlights new promising targets for the development of new metabolic drugs. Abstract: The specific molecular features of cancer cells that distinguish them from the normal ones are denoted as “hallmarks of cancer”. One of the critical hallmarks of cancer is an altered metabolism Citation: Shuvalov, O.; Daks, A.; which provides tumor cells with energy and structural resources necessary for rapid proliferation.