Microglia Monitor and Protect Neuronal Function Via Specialized Somatic Purinergic Junctions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
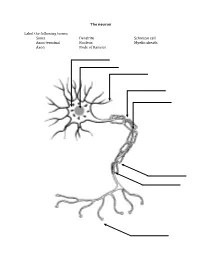
The Neuron Label the Following Terms: Soma Axon Terminal Axon Dendrite
The neuron Label the following terms: Soma Dendrite Schwaan cell Axon terminal Nucleus Myelin sheath Axon Node of Ranvier Neuron Vocabulary You must know the definitions of these terms 1. Synaptic Cleft 2. Neuron 3. Impulse 4. Sensory Neuron 5. Motor Neuron 6. Interneuron 7. Body (Soma) 8. Dendrite 9. Axon 10. Action Potential 11. Myelin Sheath (Myelin) 12. Afferent Neuron 13. Threshold 14. Neurotransmitter 15. Efferent Neurons 16. Axon Terminal 17. Stimulus 18. Refractory Period 19. Schwann 20. Nodes of Ranvier 21. Acetylcholine STEPS IN THE ACTION POTENTIAL 1. The presynaptic neuron sends neurotransmitters to postsynaptic neuron. 2. Neurotransmitters bind to receptors on the postsynaptic cell. - This action will either excite or inhibit the postsynaptic cell. - The soma becomes more positive. 3. The positive charge reaches the axon hillock. - Once the threshold of excitation is reached the neuron will fire an action potential. 4. Na+ channels open and Na+ is forced into the cell by the concentration gradient and the electrical gradient. - The neuron begins to depolarize. 5. The K+ channels open and K+ is forced out of the cell by the concentration gradient and the electrical gradient. - The neuron is depolarized. 6. The Na+ channels close at the peak of the action potential. - The neuron starts to repolarize. 7. The K+ channels close, but they close slowly and K+ leaks out. 8. The terminal buttons release neurotransmitter to the postsynaptic neuron. 9. The resting potential is overshot and the neuron falls to a -90mV (hyperpolarizes). - The neuron continues to repolarize. 10. The neuron returns to resting potential. The Synapse Label the following terms: Pre-synaptic membrane Neurotransmitters Post-synaptic membrane Synaptic cleft Vesicle Post-synaptic receptors . -

Crayfish Escape Behavior and Central Synapses
Crayfish Escape Behavior and Central Synapses. III. Electrical Junctions and Dendrite Spikes in Fast Flexor Motoneurons ROBERT S. ZUCKER Department of Biological Sciences and Program in the Neurological Sciences, Stanford University, Stanford, California 94305 THE LATERAL giant fiber is the decision fiber and fast flexor motoneurons are located in for a behavioral response in crayfish in- the dorsal neuropil of each abdominal gan- volving rapid tail flexion in response to glion. Dye injections and anatomical recon- abdominal tactile stimulation (27). Each structions of ‘identified motoneurons reveal lateral giant impulse is followed by a rapid that only those motoneurons which have tail flexion or tail flip, and when the giant dentritic branches in contact with a giant fiber does not fire in response to such stim- fiber are activated by that giant fiber (12, uli, the movement does not occur. 21). All of tl ie fast flexor motoneurons are The afferent limb of the reflex exciting excited by the ipsilateral giant; all but F4, the lateral giant has been described (28), F5 and F7 are also excited by the contra- and the habituation of the behavior has latkral giant (21 a). been explained in terms of the properties The excitation of these motoneurons, of this part of the neural circuit (29). seen as depolarizing potentials in their The properties of the neuromuscular somata, does not always reach threshold for junctions between the fast flexor motoneu- generating a spike which propagates out the rons and the phasic flexor muscles have also axon to the periphery (14). Indeed, only t,he been described (13). -

Plp-Positive Progenitor Cells Give Rise to Bergmann Glia in the Cerebellum
Citation: Cell Death and Disease (2013) 4, e546; doi:10.1038/cddis.2013.74 OPEN & 2013 Macmillan Publishers Limited All rights reserved 2041-4889/13 www.nature.com/cddis Olig2/Plp-positive progenitor cells give rise to Bergmann glia in the cerebellum S-H Chung1, F Guo2, P Jiang1, DE Pleasure2,3 and W Deng*,1,3,4 NG2 (nerve/glial antigen2)-expressing cells represent the largest population of postnatal progenitors in the central nervous system and have been classified as oligodendroglial progenitor cells, but the fate and function of these cells remain incompletely characterized. Previous studies have focused on characterizing these progenitors in the postnatal and adult subventricular zone and on analyzing the cellular and physiological properties of these cells in white and gray matter regions in the forebrain. In the present study, we examine the types of neural progeny generated by NG2 progenitors in the cerebellum by employing genetic fate mapping techniques using inducible Cre–Lox systems in vivo with two different mouse lines, the Plp-Cre-ERT2/Rosa26-EYFP and Olig2-Cre-ERT2/Rosa26-EYFP double-transgenic mice. Our data indicate that Olig2/Plp-positive NG2 cells display multipotential properties, primarily give rise to oligodendroglia but, surprisingly, also generate Bergmann glia, which are specialized glial cells in the cerebellum. The NG2 þ cells also give rise to astrocytes, but not neurons. In addition, we show that glutamate signaling is involved in distinct NG2 þ cell-fate/differentiation pathways and plays a role in the normal development of Bergmann glia. We also show an increase of cerebellar oligodendroglial lineage cells in response to hypoxic–ischemic injury, but the ability of NG2 þ cells to give rise to Bergmann glia and astrocytes remains unchanged. -

Neuregulin 1–Erbb2 Signaling Is Required for the Establishment of Radial Glia and Their Transformation Into Astrocytes in Cerebral Cortex
Neuregulin 1–erbB2 signaling is required for the establishment of radial glia and their transformation into astrocytes in cerebral cortex Ralf S. Schmid*, Barbara McGrath*, Bridget E. Berechid†, Becky Boyles*, Mark Marchionni‡, Nenad Sˇ estan†, and Eva S. Anton*§ *University of North Carolina Neuroscience Center and Department of Cell and Molecular Physiology, University of North Carolina School of Medicine, Chapel Hill, NC 27599; †Department of Neurobiology, Yale University School of Medicine, New Haven, CT 06510; and ‡CeNes Pharamceuticals, Inc., Norwood, MA 02062 Communicated by Pasko Rakic, Yale University School of Medicine, New Haven, CT, January 27, 2003 (received for review December 12, 2002) Radial glial cells and astrocytes function to support the construction mine whether NRG-1-mediated signaling is involved in radial and maintenance, respectively, of the cerebral cortex. However, the glial cell development and differentiation in the cerebral cortex. mechanisms that determine how radial glial cells are established, We show that NRG-1 signaling, involving erbB2, may act in maintained, and transformed into astrocytes in the cerebral cortex are concert with Notch signaling to exert a critical influence in the not well understood. Here, we show that neuregulin-1 (NRG-1) exerts establishment, maintenance, and appropriate transformation of a critical role in the establishment of radial glial cells. Radial glial cell radial glial cells in cerebral cortex. generation is significantly impaired in NRG mutants, and this defect can be rescued by exogenous NRG-1. Down-regulation of expression Materials and Methods and activity of erbB2, a member of the NRG-1 receptor complex, leads Clonal Analysis to Study NRG’s Role in the Initial Establishment of to the transformation of radial glial cells into astrocytes. -

Neuron Morphology Influences Axon Initial Segment Plasticity
Dartmouth College Dartmouth Digital Commons Dartmouth Scholarship Faculty Work 1-2016 Neuron Morphology Influences Axon Initial Segment Plasticity Allan T. Gulledge Dartmouth College, [email protected] Jaime J. Bravo Dartmouth College Follow this and additional works at: https://digitalcommons.dartmouth.edu/facoa Part of the Computational Neuroscience Commons, Molecular and Cellular Neuroscience Commons, and the Other Neuroscience and Neurobiology Commons Dartmouth Digital Commons Citation Gulledge, A.T. and Bravo, J.J. (2016) Neuron morphology influences axon initial segment plasticity, eNeuro doi:10.1523/ENEURO.0085-15.2016. This Article is brought to you for free and open access by the Faculty Work at Dartmouth Digital Commons. It has been accepted for inclusion in Dartmouth Scholarship by an authorized administrator of Dartmouth Digital Commons. For more information, please contact [email protected]. New Research Neuronal Excitability Neuron Morphology Influences Axon Initial Segment Plasticity1,2,3 Allan T. Gulledge1 and Jaime J. Bravo2 DOI:http://dx.doi.org/10.1523/ENEURO.0085-15.2016 1Department of Physiology and Neurobiology, Geisel School of Medicine at Dartmouth, Dartmouth-Hitchcock Medical Center, Lebanon, New Hampshire 03756, and 2Thayer School of Engineering at Dartmouth, Hanover, New Hampshire 03755 Visual Abstract In most vertebrate neurons, action potentials are initiated in the axon initial segment (AIS), a specialized region of the axon containing a high density of voltage-gated sodium and potassium channels. It has recently been proposed that neurons use plasticity of AIS length and/or location to regulate their intrinsic excitability. Here we quantify the impact of neuron morphology on AIS plasticity using computational models of simplified and realistic somatodendritic morphologies. -

Satellite Glial Cell Communication in Dorsal Root Ganglia
Neuronal somatic ATP release triggers neuron– satellite glial cell communication in dorsal root ganglia X. Zhang, Y. Chen, C. Wang, and L.-Y. M. Huang* Department of Neuroscience and Cell Biology, University of Texas Medical Branch, Galveston, TX 77555-1069 Edited by Charles F. Stevens, The Salk Institute for Biological Studies, La Jolla, CA, and approved April 25, 2007 (received for review December 14, 2006) It has been generally assumed that the cell body (soma) of a release of ATP from the somata of DRG neurons. Here, we show neuron, which contains the nucleus, is mainly responsible for that electric stimulation elicits robust vesicular release of ATP from synthesis of macromolecules and has a limited role in cell-to-cell neuronal somata and thus triggers bidirectional communication communication. Using sniffer patch recordings, we show here that between neurons and satellite cells. electrical stimulation of dorsal root ganglion (DRG) neurons elicits robust vesicular ATP release from their somata. The rate of release Results events increases with the frequency of nerve stimulation; external We asked first whether vesicular release of ATP occurs in the -Ca2؉ entry is required for the release. FM1–43 photoconversion somata of DRG neurons. P2X2-EGFP receptors were overex analysis further reveals that small clear vesicles participate in pressed in HEK cells and used as the biosensor (i.e., sniffer patch exocytosis. In addition, the released ATP activates P2X7 receptors method) (20). P2X2 receptors were chosen because they were easily in satellite cells that enwrap each DRG neuron and triggers the expressed at a high level (Ϸ200 pA/pF) in HEK cells and the communication between neuronal somata and glial cells. -

Was Not Reached, However, Even After Six to Sevenhours. A
PROTEIN SYNTHESIS IN THE ISOLATED GIANT AXON OF THE SQUID* BY A. GIUDITTA,t W.-D. DETTBARN,t AND MIROSLAv BRZIN§ MARINE BIOLOGICAL LABORATORY, WOODS HOLE, MASSACHUSETTS Communicated by David Nachmansohn, February 2, 1968 The work of Weiss and his associates,1-3 and more recently of a number of other investigators,4- has established the occurrence of a flux of materials from the soma of neurons toward the peripheral regions of the axon. It has been postulated that this mechanism would account for the origin of most of the axonal protein, although the time required to cover the distance which separates some axonal tips from their cell bodies would impose severe delays.4 On the other hand, a number of observations7-9 have indicated the occurrence of local mechanisms of synthesis in peripheral axons, as suggested by the kinetics of appearance of individual proteins after axonal transection. In this paper we report the incorporation of radioactive amino acids into the protein fraction of the axoplasm and of the axonal envelope obtained from giant axons of the squid. These axons are isolated essentially free from small fibers and connective tissue, and pure samples of axoplasm may be obtained by extru- sion of the axon. Incorporation of amino acids into axonal protein has recently been reported using systems from mammals'0 and fish."I Materials and Methods.-Giant axons of Loligo pealii were dissected and freed from small fibers: they were tied at both ends. Incubations were carried out at 18-20° in sea water previously filtered through Millipore which contained 5 mM Tris pH 7.8 and 10 Muc/ml of a mixture of 15 C'4-labeled amino acids (New England Nuclear Co., Boston, Mass.). -

Tecnicas Microscopicas
CAP 1: TÉCNICAS MICROSCÓPICAS TÉCNICAS MICROSCÓPICAS 11 Lic. Carlos R. Neira Montoya Lic. Eduardo Sedano Gelvet Lic. María Elena Vilcarromero V. El estudio de los tejidos tal como los observamos hoy en día no sería posible sin la ayuda de la histotecnología; esta disciplina se encarga del estudio de los métodos técnicas y procedimientos que permiten la transformación de un órgano en una película lo suficientemente transparente y contrastada que nos permite su observación a través del microscopio (Fig. 1-1). Para que esto ocurra se tiene que seguir una serie de pasos. Cada uno de estos pasos permite la observación de las características morfológicas del tejido que nos indica la normalidad o la alteración patológica; sin embargo en estudios mucho más minuciosos, estos pasos se harán en función de las estructuras o sustancias que se deseen investigar en la muestra correspondiente. Figura 1-1. Un órgano es transformado en una película transparente. - Pág. 5 - CAP 1: TÉCNICAS MICROSCÓPICAS Los pasos de las técnicas microscópicas para obtener un preparado histológico permanente (láminas) son: 1. Toma de la muestra. 2. Fijación. 3. Inclusión. 4. Microtomía. 5. Coloración. 1. TOMA DE LA MUESTRA Es el momento que se selecciona el órgano o tejido a estudiar. De tres fuentes puede provenir el material humano: las necropsias, las biopsias y las piezas operadas. De éstas, sólo la primera puede darnos material normal; las dos últimas habitualmente proporcionarán tejidos para estudio histopatológico. - Necropsias: son las piezas que se obtienen de un cadáver. Para histología normal es necesario que se trate de un cadáver fresco y que no haya sido atacado por ninguna lesión, por lo menos el órgano que se quiere estudiar. -

Imaging and Quantifying Ganglion Cells and Other Transparent Neurons in the Living Human Retina
Imaging and quantifying ganglion cells and other transparent neurons in the living human retina Zhuolin Liua,1, Kazuhiro Kurokawaa, Furu Zhanga, John J. Leeb, and Donald T. Millera aSchool of Optometry, Indiana University, Bloomington, IN 47405; and bPurdue School of Engineering and Technology, Indiana University–Purdue University Indianapolis, Indianapolis, IN 46202 Edited by David R. Williams, University of Rochester, Rochester, NY, and approved October 18, 2017 (received for review June 30, 2017) Ganglion cells (GCs) are fundamental to retinal neural circuitry, apoptotic GCs tagged with an intravenously administered fluores- processing photoreceptor signals for transmission to the brain via cent marker (14), thus providing direct monitoring of GC loss. The their axons. However, much remains unknown about their role in second incorporated adaptive optics (AO)—which corrects ocular vision and their vulnerability to disease leading to blindness. A aberrations—into SLO sensitive to multiply-scattered light (12). major bottleneck has been our inability to observe GCs and their This clever combination permitted imaging of a monolayer of GC degeneration in the living human eye. Despite two decades of layer (GCL) somas in areas with little or no overlying nerve fiber development of optical technologies to image cells in the living layer (NFL) (see figure 5, human result of Rossi et al.; ref. 12). By human retina, GCs remain elusive due to their high optical trans- contrast, our approach uses singly scattered light and produces lucency. Failure of conventional imaging—using predominately sin- images of unprecedented clarity of translucent retinal tissue. This gly scattered light—to reveal GCs has led to a focus on multiply- permits morphometry of GCL somas across the living human ret- scattered, fluorescence, two-photon, and phase imaging techniques ina. -

Measurements of Calcium Transients in the Soma, Neurite, and Growth Cone of Single Cultured Neurons
The Journal of Neuroscience July 1966, 6(7): 1934-1940 Measurements of Calcium Transients in the Soma, Neurite, and Growth Cone of Single Cultured Neurons Stephen R. BoIsover* and llan Spector-f *Department of Physiology, University College, London, London WClE 6BT, England, and TDepartment of Anatomical Sciences, Health Sciences Center, State University of New York at Stony Brook, Stony Brook, New York 11794 Voltage-gated changes in cytosolic free calcium ion concentra- interpret because they cannot give precise information about tion were measured in single, differentiated cells of mouse neu- the behavior of a local region of the cell or detect the presence roblastoma clone NlE-115 using the calcium-sensitive dye ar- of active calcium channels that do not contribute significantly senazo III (AIII). In cells bathed in normal medium containing to the action potential. To examine the spatial distribution of 10 mM calcium, the changes in AI11 absorbance during a single voltage-gated calcium entry in the neuronal cell membrane, we action potential indicated an increase of 1.4 nM in cytosolic used the calcium indicator dye arsenazo III (AIII) (Brown et al., calcium. When 10 mrw tetraethylammonium (TEA) was added 1975). This dye presents two important advantages. First, it can to the bath, the action potential became prolonged and the change diffuse rapidly inside the cell (R. F. Rakowski, personal com- in cytosolic calcium increased to 3.9 nM. Under these conditions, munication), thereby allowing measurements of changes in cy- repetitive stimulation at 0.5 Hz or faster caused a gradual de- tosolic calcium concentration ([Ca”],) in all regions of a single cline in the amplitude and duration of the action potential and neuron. -

Artificial Neural Networks· T\ Briet In!Toquclion
GENERAL I ARTICLE Artificial Neural Networks· t\ BrieT In!TOQuclion Jitendra R Raol and Sunilkumar S Mankame Artificial neural networks are 'biologically' inspired net works. They have the ability to learn from empirical datal information. They find use in computer science and control engineering fields. In recent years artificial neural networks (ANNs) have fascinated scientists and engineers all over the world. They have the ability J R Raol is a scientist with to learn and recall - the main functions of the (human) brain. A NAL. His research major reason for this fascination is that ANNs are 'biologically' interests are parameter inspired. They have the apparent ability to imitate the brain's estimation, neural networks, fuzzy systems, activity to make decisions and draw conclusions when presented genetic algorithms and with complex and noisy information. However there are vast their applications to differences between biological neural networks (BNNs) of the aerospace problems. He brain and ANN s. writes poems in English. A thorough understanding of biologically derived NNs requires knowledge from other sciences: biology, mathematics and artifi cial intelligence. However to understand the basics of ANNs, a knowledge of neurobiology is not necessary. Yet, it is a good idea to understand how ANNs have been derived from real biological neural systems (see Figures 1,2 and the accompanying boxes). The soma of the cell body receives inputs from other neurons via Sunilkumar S Mankame is adaptive synaptic connections to the dendrites and when a neuron a graduate research student from Regional is excited, the nerve impulses from the soma are transmitted along Engineering College, an axon to the synapses of other neurons. -

Oligodendroglial Energy Metabolism and (Re)Myelination
life Review Oligodendroglial Energy Metabolism and (re)Myelination Vanja Tepavˇcevi´c Achucarro Basque Center for Neuroscience, University of the Basque Country, Parque Cientifico de la UPV/EHU, Barrio Sarriena s/n, Edificio Sede, Planta 3, 48940 Leioa, Spain; [email protected] Abstract: Central nervous system (CNS) myelin has a crucial role in accelerating the propagation of action potentials and providing trophic support to the axons. Defective myelination and lack of myelin regeneration following demyelination can both lead to axonal pathology and neurode- generation. Energy deficit has been evoked as an important contributor to various CNS disorders, including multiple sclerosis (MS). Thus, dysregulation of energy homeostasis in oligodendroglia may be an important contributor to myelin dysfunction and lack of repair observed in the disease. This article will focus on energy metabolism pathways in oligodendroglial cells and highlight differences dependent on the maturation stage of the cell. In addition, it will emphasize that the use of alternative energy sources by oligodendroglia may be required to save glucose for functions that cannot be fulfilled by other metabolites, thus ensuring sufficient energy input for both myelin synthesis and trophic support to the axons. Finally, it will point out that neuropathological findings in a subtype of MS lesions likely reflect defective oligodendroglial energy homeostasis in the disease. Keywords: energy metabolism; oligodendrocyte; oligodendrocyte progenitor cell; myelin; remyeli- nation; multiple sclerosis; glucose; ketone bodies; lactate; N-acetyl aspartate Citation: Tepavˇcevi´c,V. Oligodendroglial Energy Metabolism 1. Introduction and (re)Myelination. Life 2021, 11, Myelination is the key evolutionary event in the development of higher vertebrates. 238.