Oligodendroglial Energy Metabolism and (Re)Myelination
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Screening and Identification of Key Biomarkers in Clear Cell Renal Cell Carcinoma Based on Bioinformatics Analysis
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Screening and identification of key biomarkers in clear cell renal cell carcinoma based on bioinformatics analysis Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Clear cell renal cell carcinoma (ccRCC) is one of the most common types of malignancy of the urinary system. The pathogenesis and effective diagnosis of ccRCC have become popular topics for research in the previous decade. In the current study, an integrated bioinformatics analysis was performed to identify core genes associated in ccRCC. An expression dataset (GSE105261) was downloaded from the Gene Expression Omnibus database, and included 26 ccRCC and 9 normal kideny samples. Assessment of the microarray dataset led to the recognition of differentially expressed genes (DEGs), which was subsequently used for pathway and gene ontology (GO) enrichment analysis. -
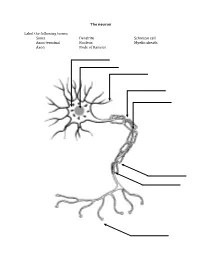
The Neuron Label the Following Terms: Soma Axon Terminal Axon Dendrite
The neuron Label the following terms: Soma Dendrite Schwaan cell Axon terminal Nucleus Myelin sheath Axon Node of Ranvier Neuron Vocabulary You must know the definitions of these terms 1. Synaptic Cleft 2. Neuron 3. Impulse 4. Sensory Neuron 5. Motor Neuron 6. Interneuron 7. Body (Soma) 8. Dendrite 9. Axon 10. Action Potential 11. Myelin Sheath (Myelin) 12. Afferent Neuron 13. Threshold 14. Neurotransmitter 15. Efferent Neurons 16. Axon Terminal 17. Stimulus 18. Refractory Period 19. Schwann 20. Nodes of Ranvier 21. Acetylcholine STEPS IN THE ACTION POTENTIAL 1. The presynaptic neuron sends neurotransmitters to postsynaptic neuron. 2. Neurotransmitters bind to receptors on the postsynaptic cell. - This action will either excite or inhibit the postsynaptic cell. - The soma becomes more positive. 3. The positive charge reaches the axon hillock. - Once the threshold of excitation is reached the neuron will fire an action potential. 4. Na+ channels open and Na+ is forced into the cell by the concentration gradient and the electrical gradient. - The neuron begins to depolarize. 5. The K+ channels open and K+ is forced out of the cell by the concentration gradient and the electrical gradient. - The neuron is depolarized. 6. The Na+ channels close at the peak of the action potential. - The neuron starts to repolarize. 7. The K+ channels close, but they close slowly and K+ leaks out. 8. The terminal buttons release neurotransmitter to the postsynaptic neuron. 9. The resting potential is overshot and the neuron falls to a -90mV (hyperpolarizes). - The neuron continues to repolarize. 10. The neuron returns to resting potential. The Synapse Label the following terms: Pre-synaptic membrane Neurotransmitters Post-synaptic membrane Synaptic cleft Vesicle Post-synaptic receptors . -

Mapping Influenza-Induced Posttranslational Modifications On
viruses Article Mapping Influenza-Induced Posttranslational Modifications on Histones from CD8+ T Cells Svetlana Rezinciuc 1, Zhixin Tian 2, Si Wu 2, Shawna Hengel 2, Ljiljana Pasa-Tolic 2 and Heather S. Smallwood 1,3,* 1 Department of Pediatrics, University of Tennessee Health Science Center, Memphis, TN 38163, USA; [email protected] 2 Environmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, Richland, WA 99354, USA; [email protected] (Z.T.); [email protected] (S.W.); [email protected] (S.H.); [email protected] (L.P.-T.) 3 Children’s Foundation Research Institute, Memphis, TN 38105, USA * Correspondence: [email protected]; Tel.: +1-(901)-448–3068 Academic Editor: Italo Tempera Received: 10 October 2020; Accepted: 2 December 2020; Published: 8 December 2020 Abstract: T cell function is determined by transcriptional networks that are regulated by epigenetic programming via posttranslational modifications (PTMs) to histone proteins and DNA. Bottom-up mass spectrometry (MS) can identify histone PTMs, whereas intact protein analysis by MS can detect species missed by bottom-up approaches. We used a novel approach of online two-dimensional liquid chromatography-tandem MS with high-resolution reversed-phase liquid chromatography (RPLC), alternating electron transfer dissociation (ETD) and collision-induced dissociation (CID) on precursor ions to maximize fragmentation of uniquely modified species. The first online RPLC separation sorted histone families, then RPLC or weak cation exchange hydrophilic interaction liquid chromatography (WCX-HILIC) separated species heavily clad in PTMs. Tentative identifications were assigned by matching proteoform masses to predicted theoretical masses that were verified with tandem MS. We used this innovative approach for histone-intact protein PTM mapping (HiPTMap) to identify and quantify proteoforms purified from CD8 T cells after in vivo influenza infection. -

Crayfish Escape Behavior and Central Synapses
Crayfish Escape Behavior and Central Synapses. III. Electrical Junctions and Dendrite Spikes in Fast Flexor Motoneurons ROBERT S. ZUCKER Department of Biological Sciences and Program in the Neurological Sciences, Stanford University, Stanford, California 94305 THE LATERAL giant fiber is the decision fiber and fast flexor motoneurons are located in for a behavioral response in crayfish in- the dorsal neuropil of each abdominal gan- volving rapid tail flexion in response to glion. Dye injections and anatomical recon- abdominal tactile stimulation (27). Each structions of ‘identified motoneurons reveal lateral giant impulse is followed by a rapid that only those motoneurons which have tail flexion or tail flip, and when the giant dentritic branches in contact with a giant fiber does not fire in response to such stim- fiber are activated by that giant fiber (12, uli, the movement does not occur. 21). All of tl ie fast flexor motoneurons are The afferent limb of the reflex exciting excited by the ipsilateral giant; all but F4, the lateral giant has been described (28), F5 and F7 are also excited by the contra- and the habituation of the behavior has latkral giant (21 a). been explained in terms of the properties The excitation of these motoneurons, of this part of the neural circuit (29). seen as depolarizing potentials in their The properties of the neuromuscular somata, does not always reach threshold for junctions between the fast flexor motoneu- generating a spike which propagates out the rons and the phasic flexor muscles have also axon to the periphery (14). Indeed, only t,he been described (13). -

• Our Bodies Make All the Cholesterol We Need. • 85 % of Our Blood
• Our bodies make all the cholesterol we need. • 85 % of our blood cholesterol level is endogenous • 15 % = dietary from meat, poultry, fish, seafood and dairy products. • It's possible for some people to eat foods high in cholesterol and still have low blood cholesterol levels. • Likewise, it's possible to eat foods low in cholesterol and have a high blood cholesterol level SYNTHESIS OF CHOLESTEROL • LOCATION • All tissues • Liver • Cortex of adrenal gland • Gonads • Smooth endoplasmic reticulum Cholesterol biosynthesis and degradation • Diet: only found in animal fat • Biosynthesis: primarily synthesized in the liver from acetyl-coA; biosynthesis is inhibited by LDL uptake • Degradation: only occurs in the liver • Cholesterol is only synthesized by animals • Although de novo synthesis of cholesterol occurs in/ by almost all tissues in humans, the capacity is greatest in liver, intestine, adrenal cortex, and reproductive tissues, including ovaries, testes, and placenta. • Most de novo synthesis occurs in the liver, where cholesterol is synthesized from acetyl-CoA in the cytoplasm. • Biosynthesis in the liver accounts for approximately 10%, and in the intestines approximately 15%, of the amount produced each day. • Since cholesterol is not synthesized in plants; vegetables & fruits play a major role in low cholesterol diets. • As previously mentioned, cholesterol biosynthesis is necessary for membrane synthesis, and as a precursor for steroid synthesis including steroid hormone and vitamin D production, and bile acid synthesis, in the liver. • Slightly less than half of the cholesterol in the body derives from biosynthesis de novo. • Most cells derive their cholesterol from LDL or HDL, but some cholesterol may be synthesize: de novo. -

Cholesterol Metabolites 25-Hydroxycholesterol and 25-Hydroxycholesterol 3-Sulfate Are Potent Paired Regulators: from Discovery to Clinical Usage
H OH metabolites OH Review Cholesterol Metabolites 25-Hydroxycholesterol and 25-Hydroxycholesterol 3-Sulfate Are Potent Paired Regulators: From Discovery to Clinical Usage Yaping Wang 1, Xiaobo Li 2 and Shunlin Ren 1,* 1 Department of Internal Medicine, McGuire Veterans Affairs Medical Center, Virginia Commonwealth University, Richmond, VA 23249, USA; [email protected] 2 Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Fudan University, Shanghai 200032, China; [email protected] * Correspondence: [email protected]; Tel.: +1-(804)-675-5000 (ext. 4973) Abstract: Oxysterols have long been believed to be ligands of nuclear receptors such as liver × recep- tor (LXR), and they play an important role in lipid homeostasis and in the immune system, where they are involved in both transcriptional and posttranscriptional mechanisms. However, they are increas- ingly associated with a wide variety of other, sometimes surprising, cell functions. Oxysterols have also been implicated in several diseases such as metabolic syndrome. Oxysterols can be sulfated, and the sulfated oxysterols act in different directions: they decrease lipid biosynthesis, suppress inflammatory responses, and promote cell survival. Our recent reports have shown that oxysterol and oxysterol sulfates are paired epigenetic regulators, agonists, and antagonists of DNA methyl- transferases, indicating that their function of global regulation is through epigenetic modification. In this review, we explore our latest research of 25-hydroxycholesterol and 25-hydroxycholesterol 3-sulfate in a novel regulatory mechanism and evaluate the current evidence for these roles. Citation: Wang, Y.; Li, X.; Ren, S. Keywords: oxysterol sulfates; oxysterol sulfation; epigenetic regulators; 25-hydroxysterol; Cholesterol Metabolites 25-hydroxycholesterol 3-sulfate; 25-hydroxycholesterol 3,25-disulfate 25-Hydroxycholesterol and 25-Hydroxycholesterol 3-Sulfate Are Potent Paired Regulators: From Discovery to Clinical Usage. -

Plp-Positive Progenitor Cells Give Rise to Bergmann Glia in the Cerebellum
Citation: Cell Death and Disease (2013) 4, e546; doi:10.1038/cddis.2013.74 OPEN & 2013 Macmillan Publishers Limited All rights reserved 2041-4889/13 www.nature.com/cddis Olig2/Plp-positive progenitor cells give rise to Bergmann glia in the cerebellum S-H Chung1, F Guo2, P Jiang1, DE Pleasure2,3 and W Deng*,1,3,4 NG2 (nerve/glial antigen2)-expressing cells represent the largest population of postnatal progenitors in the central nervous system and have been classified as oligodendroglial progenitor cells, but the fate and function of these cells remain incompletely characterized. Previous studies have focused on characterizing these progenitors in the postnatal and adult subventricular zone and on analyzing the cellular and physiological properties of these cells in white and gray matter regions in the forebrain. In the present study, we examine the types of neural progeny generated by NG2 progenitors in the cerebellum by employing genetic fate mapping techniques using inducible Cre–Lox systems in vivo with two different mouse lines, the Plp-Cre-ERT2/Rosa26-EYFP and Olig2-Cre-ERT2/Rosa26-EYFP double-transgenic mice. Our data indicate that Olig2/Plp-positive NG2 cells display multipotential properties, primarily give rise to oligodendroglia but, surprisingly, also generate Bergmann glia, which are specialized glial cells in the cerebellum. The NG2 þ cells also give rise to astrocytes, but not neurons. In addition, we show that glutamate signaling is involved in distinct NG2 þ cell-fate/differentiation pathways and plays a role in the normal development of Bergmann glia. We also show an increase of cerebellar oligodendroglial lineage cells in response to hypoxic–ischemic injury, but the ability of NG2 þ cells to give rise to Bergmann glia and astrocytes remains unchanged. -

Genetic Deletion of Abcc6 Disturbs Cholesterol Homeostasis in Mice Bettina Ibold1, Janina Tiemann1, Isabel Faust1, Uta Ceglarek2, Julia Dittrich2, Theo G
www.nature.com/scientificreports OPEN Genetic deletion of Abcc6 disturbs cholesterol homeostasis in mice Bettina Ibold1, Janina Tiemann1, Isabel Faust1, Uta Ceglarek2, Julia Dittrich2, Theo G. M. F. Gorgels3,4, Arthur A. B. Bergen4,5, Olivier Vanakker6, Matthias Van Gils6, Cornelius Knabbe1 & Doris Hendig1* Genetic studies link adenosine triphosphate-binding cassette transporter C6 (ABCC6) mutations to pseudoxanthoma elasticum (PXE). ABCC6 sequence variations are correlated with altered HDL cholesterol levels and an elevated risk of coronary artery diseases. However, the role of ABCC6 in cholesterol homeostasis is not widely known. Here, we report reduced serum cholesterol and phytosterol levels in Abcc6-defcient mice, indicating an impaired sterol absorption. Ratios of cholesterol precursors to cholesterol were increased, confrmed by upregulation of hepatic 3-hydroxy-3-methylglutaryl coenzyme A reductase (Hmgcr) expression, suggesting activation of cholesterol biosynthesis in Abcc6−/− mice. We found that cholesterol depletion was accompanied by a substantial decrease in HDL cholesterol mediated by lowered ApoA-I and ApoA-II protein levels and not by inhibited lecithin-cholesterol transferase activity. Additionally, higher proprotein convertase subtilisin/kexin type 9 (Pcsk9) serum levels in Abcc6−/− mice and PXE patients and elevated ApoB level in knockout mice were observed, suggesting a potentially altered very low-density lipoprotein synthesis. Our results underline the role of Abcc6 in cholesterol homeostasis and indicate impaired cholesterol metabolism as an important pathomechanism involved in PXE manifestation. Mutations in the adenosine triphosphate-binding cassette transporter C6 (ABCC6) gene are responsible for pseudoxanthoma elasticum (PXE), a metabolic disease, hallmarked by a progressive elastic fber calcifcation of the skin, eyes and cardiovascular system. -

Exogenous Hydrogen Sulfide Plays an Important Role by Regulating
International Journal of Molecular Sciences Review Exogenous Hydrogen Sulfide Plays an Important Role by Regulating Autophagy in Diabetic-Related Diseases Shuangyu Lv , Huiyang Liu and Honggang Wang * Henan International Joint Laboratory of Nuclear Protein Regulation, School of Basic Medical Sciences, Henan University, Kaifeng 475000, China; [email protected] (S.L.); [email protected] (H.L.) * Correspondence: [email protected] Abstract: Autophagy is a vital cell mechanism which plays an important role in many physiological processes including clearing long-lived, accumulated and misfolded proteins, removing damaged organelles and regulating growth and aging. Autophagy also participates in a variety of biological functions, such as development, cell differentiation, resistance to pathogens and nutritional hunger. Recently, autophagy has been reported to be involved in diabetes, but the mechanism is not fully understood. Hydrogen sulfide (H2S) is a colorless, water-soluble, flammable gas with the typical odor of rotten eggs, which has been known as a highly toxic gas for many years. However, it has been reported recently that H2S, together with nitric oxide and carbon monoxide, is an important gas signal transduction molecule. H2S has been reported to play a protective role in many diabetes- related diseases, but the mechanism is not fully clear. Recent studies indicate that H2S plays an important role by regulating autophagy in many diseases including cancer, tissue fibrosis diseases and glycometabolic diseases; however, the related mechanism has not been fully studied. In this review, we summarize recent research on the role of H2S in regulating autophagy in diabetic-related diseases to provide references for future related research. -

Neuregulin 1–Erbb2 Signaling Is Required for the Establishment of Radial Glia and Their Transformation Into Astrocytes in Cerebral Cortex
Neuregulin 1–erbB2 signaling is required for the establishment of radial glia and their transformation into astrocytes in cerebral cortex Ralf S. Schmid*, Barbara McGrath*, Bridget E. Berechid†, Becky Boyles*, Mark Marchionni‡, Nenad Sˇ estan†, and Eva S. Anton*§ *University of North Carolina Neuroscience Center and Department of Cell and Molecular Physiology, University of North Carolina School of Medicine, Chapel Hill, NC 27599; †Department of Neurobiology, Yale University School of Medicine, New Haven, CT 06510; and ‡CeNes Pharamceuticals, Inc., Norwood, MA 02062 Communicated by Pasko Rakic, Yale University School of Medicine, New Haven, CT, January 27, 2003 (received for review December 12, 2002) Radial glial cells and astrocytes function to support the construction mine whether NRG-1-mediated signaling is involved in radial and maintenance, respectively, of the cerebral cortex. However, the glial cell development and differentiation in the cerebral cortex. mechanisms that determine how radial glial cells are established, We show that NRG-1 signaling, involving erbB2, may act in maintained, and transformed into astrocytes in the cerebral cortex are concert with Notch signaling to exert a critical influence in the not well understood. Here, we show that neuregulin-1 (NRG-1) exerts establishment, maintenance, and appropriate transformation of a critical role in the establishment of radial glial cells. Radial glial cell radial glial cells in cerebral cortex. generation is significantly impaired in NRG mutants, and this defect can be rescued by exogenous NRG-1. Down-regulation of expression Materials and Methods and activity of erbB2, a member of the NRG-1 receptor complex, leads Clonal Analysis to Study NRG’s Role in the Initial Establishment of to the transformation of radial glial cells into astrocytes. -

Subcellular Distribution of HDAC1 in Neurotoxic Conditions Is Dependent on Serine Phosphorylation
The Journal of Neuroscience, August 2, 2017 • 37(31):7547–7559 • 7547 Neurobiology of Disease Subcellular Distribution of HDAC1 in Neurotoxic Conditions Is Dependent on Serine Phosphorylation Yunjiao Zhu,1* Oscar G. Vidaurre,1* XKadidia P. Adula,1 XNebojsa Kezunovic,1 XMaureen Wentling,1 X George W. Huntley,1 and XPatrizia Casaccia1,2 1Department of Neuroscience, Friedman Brain Institute, Graduate School of Biomedical Sciences, Icahn School of Medicine at Mount Sinai, New York, New York 10029, and 2Advanced Science Research Center at the Graduate Center of the City University of New York, New York, New York 10031 Calcium-dependent nuclear export of histone deacetylase 1 (HDAC1) was shown previously to precede axonal damage in culture, but the in vivo relevance of these findings and the potential posttranslational modifications of HDAC1 remained elusive. Using acute hippocam- pal slices from mice of either sex with genetic conditional ablation of Hdac1 in CA1 hippocampal neurons (i.e., Camk2a-cre;Hdac1 fl/fl), we show significantly diminished axonal damage in response to neurotoxic stimuli. The protective effect of Hdac1 ablation was detected also in CA3 neurons in Grik4-cre;Hdac1 fl/f mice, which were more resistant to the excitotoxic damage induced by intraventricular injection of kainic acid. The amino acid residues modulating HDAC1 subcellular localization were identified by site-directed mutagenesis, which identified serine residues 421 and 423 as critical for its nuclear localization. The physiological phosphorylation of HDAC1 was decreased by neurotoxic stimuli, which stimulated the phosphatase enzymatic activity of calcineurin. Treatment of neurons with the calcineurin inhibitors FK506 or cyclosporin A resulted in nuclear accumulation of phospho-HDAC1 and was neuroprotective. -

ASPA Gene Aspartoacylase
ASPA gene aspartoacylase Normal Function The ASPA gene provides instructions for making an enzyme called aspartoacylase. In the brain, this enzyme breaks down a compound called N-acetyl-L-aspartic acid (NAA) into aspartic acid (an amino acid that is a building block of many proteins) and another molecule called acetic acid. The production and breakdown of NAA appears to be critical for maintaining the brain's white matter, which consists of nerve fibers surrounded by a myelin sheath. The myelin sheath is the covering that protects nerve fibers and promotes the efficient transmission of nerve impulses. The precise function of NAA is unclear. Researchers had suspected that it played a role in the production of the myelin sheath, but recent studies suggest that NAA does not have this function. The enzyme may instead be involved in the transport of water molecules out of nerve cells (neurons). Health Conditions Related to Genetic Changes Canavan disease More than 80 mutations in the ASPA gene are known to cause Canavan disease, which is a rare inherited disorder that affects brain development. Researchers have described two major forms of this condition: neonatal/infantile Canavan disease, which is the most common and most severe form, and mild/juvenile Canavan disease. The ASPA gene mutations that cause the neonatal/infantile form severely impair the activity of aspartoacylase, preventing the breakdown of NAA and allowing this substance to build up to high levels in the brain. The mutations that cause the mild/juvenile form have milder effects on the enzyme's activity, leading to less accumulation of NAA.