Investigations Into Ambient Ionization Mechanisms: Electrospray Ionization and Direct Analysis in Real Time Mass Spectrometry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Multiplatform Investigation of Plasma and Tissue Lipid Signatures of Breast Cancer Using Mass Spectrometry Tools
International Journal of Molecular Sciences Article Multiplatform Investigation of Plasma and Tissue Lipid Signatures of Breast Cancer Using Mass Spectrometry Tools 1, 2, 2 3 Alex Ap. Rosini Silva y, Marcella R. Cardoso y , Luciana Montes Rezende , John Q. Lin , Fernando Guimaraes 2 , Geisilene R. Paiva Silva 4, Michael Murgu 5, Denise Gonçalves Priolli 1, Marcos N. Eberlin 6, Alessandra Tata 7, Livia S. Eberlin 3 , Sophie F. M. Derchain 2 and Andreia M. Porcari 1,* 1 Postgraduate Program of Health Sciences, São Francisco University, Bragança Paulista SP 12916-900, Brazil; [email protected] (A.A.R.S.); [email protected] (D.G.P.) 2 Department of Gynecological and Breast Oncology, Women’s Hospital (CAISM), Faculty of Medical Sciences, State University of Campinas (UNICAMP), Campinas SP 13083-881, Brazil; [email protected] (M.R.C.); [email protected] (L.M.R.); [email protected] (F.G.); [email protected] (S.F.M.D.) 3 Department of Chemistry, The University of Texas at Austin, Austin, TX 78712, USA; [email protected] (J.Q.L.); [email protected] (L.S.E.) 4 Laboratory of Molecular and Investigative Pathology—LAPE, Women’s Hospital (CAISM), Faculty of Medical Sciences, State University of Campinas (UNICAMP), Campinas SP 13083-881, Brazil; [email protected] 5 Waters Corporation, São Paulo, SP 13083-970, Brazil; [email protected] 6 School of Engineering, Mackenzie Presbyterian University, São Paulo SP 01302-907, Brazil; [email protected] 7 Laboratorio di Chimica Sperimentale, Istituto Zooprofilattico Sperimentale delle Venezie, Viale Fiume 78, 36100 Vicenza, Italy; [email protected] * Correspondence: [email protected]; Tel.: +55-11-2454-8047 These authors contributed equally to this work. -

Contents • Introduction to Proteomics and Mass Spectrometry
Contents • Introduction to Proteomics and Mass spectrometry - What we need to know in our quest to explain life - Fundamental things you need to know about mass spectrometry • Interfaces and ion sources - Electrospray ionization (ESI) - conventional and nanospray - Heated nebulizer atmospheric pressure chemical ionization - Matrix assisted laser desorption • Types of MS analyzers - Magnetic sector - Quadrupole - Time-of-flight - Hybrid - Ion trap In the next step in our quest to explain what is life The human genome project has largely been completed and many other genomes are surrendering to the gene sequencers. However, all this knowledge does not give us the information that is needed to explain how living cells work. To do that, we need to study proteins. In 2002, mass spectrometry has developed to the point where it has the capacity to obtain the "exact" molecular weight of many macromolecules. At the present time, this includes proteins up to 150,000 Da. Proteins of higher molecular weights (up to 500,000 Da) can also be studied by mass spectrometry, but with less accuracy. The paradigm for sequencing of peptides and identification of proteins has changed – because of the availability of the human genome database, peptides can be identified merely by their masses or by partial sequence information, often in minutes, not hours. This new capacity is shifting the emphasis of biomedical research back to the functional aspects of cell biochemistry, the expression of particular sets of genes and their gene products, the proteins of the cell. These are the new goals of the biological scientist: o to know which proteins are expressed in each cell, preferably one cell at a time o to know how these proteins are modified, information that cannot necessarily be deduced from the nucleotide sequence of individual genes. -

Electron Ionization
Chapter 6 Chapter 6 Electron Ionization I. Introduction ......................................................................................................317 II. Ionization Process............................................................................................317 III. Strategy for Data Interpretation......................................................................321 1. Assumptions 2. The Ionization Process IV. Types of Fragmentation Pathways.................................................................328 1. Sigma-Bond Cleavage 2. Homolytic or Radical-Site-Driven Cleavage 3. Heterolytic or Charge-Site-Driven Cleavage 4. Rearrangements A. Hydrogen-Shift Rearrangements B. Hydride-Shift Rearrangements V. Representative Fragmentations (Spectra) of Classes of Compounds.......... 344 1. Hydrocarbons A. Saturated Hydrocarbons 1) Straight-Chain Hydrocarbons 2) Branched Hydrocarbons 3) Cyclic Hydrocarbons B. Unsaturated C. Aromatic 2. Alkyl Halides 3. Oxygen-Containing Compounds A. Aliphatic Alcohols B. Aliphatic Ethers C. Aromatic Alcohols D. Cyclic Ethers E. Ketones and Aldehydes F. Aliphatic Acids and Esters G. Aromatic Acids and Esters 4. Nitrogen-Containing Compounds A. Aliphatic Amines B. Aromatic Compounds Containing Atoms of Nitrogen C. Heterocyclic Nitrogen-Containing Compounds D. Nitro Compounds E. Concluding Remarks on the Mass Spectra of Nitrogen-Containing Compounds 5. Multiple Heteroatoms or Heteroatoms and a Double Bond 6. Trimethylsilyl Derivative 7. Determining the Location of Double Bonds VI. Library -

Modern Mass Spectrometry
Modern Mass Spectrometry MacMillan Group Meeting 2005 Sandra Lee Key References: E. Uggerud, S. Petrie, D. K. Bohme, F. Turecek, D. Schröder, H. Schwarz, D. Plattner, T. Wyttenbach, M. T. Bowers, P. B. Armentrout, S. A. Truger, T. Junker, G. Suizdak, Mark Brönstrup. Topics in Current Chemistry: Modern Mass Spectroscopy, pp. 1-302, 225. Springer-Verlag, Berlin, 2003. Current Topics in Organic Chemistry 2003, 15, 1503-1624 1 The Basics of Mass Spectroscopy ! Purpose Mass spectrometers use the difference in mass-to-charge ratio (m/z) of ionized atoms or molecules to separate them. Therefore, mass spectroscopy allows quantitation of atoms or molecules and provides structural information by the identification of distinctive fragmentation patterns. The general operation of a mass spectrometer is: "1. " create gas-phase ions "2. " separate the ions in space or time based on their mass-to-charge ratio "3. " measure the quantity of ions of each mass-to-charge ratio Ionization sources ! Instrumentation Chemical Ionisation (CI) Atmospheric Pressure CI!(APCI) Electron Impact!(EI) Electrospray Ionization!(ESI) SORTING DETECTION IONIZATION OF IONS OF IONS Fast Atom Bombardment (FAB) Field Desorption/Field Ionisation (FD/FI) Matrix Assisted Laser Desorption gaseous mass ion Ionisation!(MALDI) ion source analyzer transducer Thermospray Ionisation (TI) Analyzers quadrupoles vacuum signal Time-of-Flight (TOF) pump processor magnetic sectors 10-5– 10-8 torr Fourier transform and quadrupole ion traps inlet Detectors mass electron multiplier spectrum Faraday cup Ionization Sources: Classical Methods ! Electron Impact Ionization A beam of electrons passes through a gas-phase sample and collides with neutral analyte molcules (M) to produce a positively charged ion or a fragment ion. -
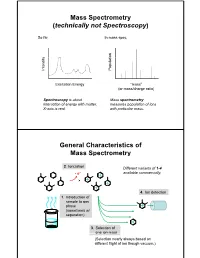
Mass Spectrometry (Technically Not Spectroscopy)
Mass Spectrometry (technically not Spectroscopy) So far, In mass spec, on y Populati Intensit Excitation Energy “mass” (or mass/charge ratio) Spectroscopy is about Mass spectrometry interaction of energy with matter. measures population of ions X-axis is real. with particular mass. General Characteristics of Mass Spectrometry 2. Ionization Different variants of 1-4 -e- available commercially. 4. Ion detection 1. Introduction of sample to gas phase (sometimes w/ separation) 3. Selection of one ion mass (Selection nearly always based on different flight of ion though vacuum.) General Components of a Mass Spectrometer Lots of choices, which can be mixed and matched. direct injection The Mass Spectrum fragment “daughter” ions M+ “parent” mass Sample Introduction: Direc t Inser tion Prob e If sample is a liquid, sample can also be injected directly into ionization region. If sample isn’t pure, get multiple parents (that can’t be distinguished from fragments). Capillary Column Introduction Continous source of molecules to spectrometer. detector column (including GC, LC, chiral, size exclusion) • Signal intensity depends on both amount of molecule and ionization efficiency • To use quantitatively, must calibrate peaks with respect eltilution time ttlitotal ion curren t to quantity eluted (TIC) over time Capillary Column Introduction Easy to interface with gas or liquid chromatography. TIC trace elution time time averaged time averaged mass spectrum mass spectrum Methods of Ionization: Electron Ionization (EI) 1 - + - 1 M + e (kV energy) M + 2e Fragmentation in Electron Ionization daughter ion (observed in spectrum) neutral fragment (not observed) excited parent at electron at electron energy of energy of 15 eV 70 e V Lower electron energy yields less fragmentation, but also less signal. -

Portable Mass Spectrometry
Food Analytical Methods https://doi.org/10.1007/s12161-019-01666-6 Potential of Recent Ambient Ionization Techniques for Future Food Contaminant Analysis Using (Trans)Portable Mass Spectrometry Marco H. Blokland1 & Arjen Gerssen1 & Paul W. Zoontjes1 & Janusz Pawliszyn2 & Michel W.F. Nielen1,3 Received: 3 June 2019 /Accepted: 31 October 2019 # The Author(s) 2019 Abstract In food analysis, a trend towards on-site testing of quality and safety parameters is emerging. So far, on-site testing has been mainly explored by miniaturized optical spectroscopy and ligand-binding assay approaches such as lateral flow immunoassays and biosensors. However, for the analysis of multiple parameters at regulatory levels, mass spectrometry (MS) is the method of choice in food testing laboratories. Thanks to recent developments in ambient ionization and upcoming miniaturization of mass analyzers, (trans)portable mass spectrometry may be added to the toolkit for on-site testing and eventually compete with multiplex immunoassays in mixture analysis. In this study, we preliminary evaluated a selection of recent ambient ionization techniques for their potential in simplified testing of selected food contaminants such as pesticides, veterinary drugs, and natural toxins, aiming for a minimum in sample preparation while maintaining acceptable sensitivity and robustness. Matrix-assisted inlet ionization (MAI), handheld desorption atmospheric pressure chemical ionization (DAPCI), transmission-mode direct analysis in real time (TM-DART), and coated blade spray (CBS) were coupled to both benchtop Orbitrap and compact quad- rupole single-stage mass analyzers, while CBS was also briefly studied on a benchtop triple-quadrupole MS. From the results, it can be concluded that for solid and liquid sample transmission configurations provide the highest sensitivity while upon addition ofastationaryphase,suchasinCBS,evenlowμg/L levels in urine samples can be achieved provided the additional selectivity of tandem mass spectrometry is exploited. -

An Introduction to Mass Spectrometry
An Introduction to Mass Spectrometry by Scott E. Van Bramer Widener University Department of Chemistry One University Place Chester, PA 19013 [email protected] http://science.widener.edu/~svanbram revised: September 2, 1998 © Copyright 1997 TABLE OF CONTENTS INTRODUCTION ........................................................... 4 SAMPLE INTRODUCTION ....................................................5 Direct Vapor Inlet .......................................................5 Gas Chromatography.....................................................5 Liquid Chromatography...................................................6 Direct Insertion Probe ....................................................6 Direct Ionization of Sample ................................................6 IONIZATION TECHNIQUES...................................................6 Electron Ionization .......................................................7 Chemical Ionization ..................................................... 9 Fast Atom Bombardment and Secondary Ion Mass Spectrometry .................10 Atmospheric Pressure Ionization and Electrospray Ionization ....................11 Matrix Assisted Laser Desorption/Ionization ................................ 13 Other Ionization Methods ................................................13 Self-Test #1 ...........................................................14 MASS ANALYZERS .........................................................14 Quadrupole ............................................................15 -

An Organic Chemist's Guide to Electrospray Mass Spectrometric
molecules Review An Organic Chemist’s Guide to Electrospray Mass Spectrometric Structure Elucidation Arnold Steckel 1 and Gitta Schlosser 2,* 1 Hevesy György PhD School of Chemistry, ELTE Eötvös Loránd University, Pázmány Péter sétány 1/A, 1117 Budapest, Hungary; [email protected] 2 Department of Analytical Chemistry, ELTE Eötvös Loránd University, Pázmány Péter sétány 1/A, 1117 Budapest, Hungary * Correspondence: [email protected] Received: 16 January 2019; Accepted: 8 February 2019; Published: 10 February 2019 Abstract: Tandem mass spectrometry is an important tool for structure elucidation of natural and synthetic organic products. Fragmentation of odd electron ions (OE+) generated by electron ionization (EI) was extensively studied in the last few decades, however there are only a few systematic reviews available concerning the fragmentation of even-electron ions (EE+/EE−) produced by the currently most common ionization techniques, electrospray ionization (ESI) and atmospheric pressure chemical ionization (APCI). This review summarizes the most important features of tandem mass spectra generated by collision-induced dissociation fragmentation and presents didactic examples for the unexperienced users. Keywords: tandem mass spectrometry; MS/MS fragmentation; collision-induced dissociation; CID; ESI; structure elucidation 1. Introduction Electron ionization (EI), a hard ionization technique, is the method of choice for analyses of small (<1000 Da), nonpolar, volatile compounds. As its name implies, the technique involves ionization by electrons with ~70 eV energy. This energy is high enough to yield very reproducible mass spectra with a large number of fragments. However, these spectra frequently lack the radical type molecular ions (M+) due to the high internal energy transferred to the precursors [1]. -

Methods of Ion Generation
Chem. Rev. 2001, 101, 361−375 361 Methods of Ion Generation Marvin L. Vestal PE Biosystems, Framingham, Massachusetts 01701 Received May 24, 2000 Contents I. Introduction 361 II. Atomic Ions 362 A. Thermal Ionization 362 B. Spark Source 362 C. Plasma Sources 362 D. Glow Discharge 362 E. Inductively Coupled Plasma (ICP) 363 III. Molecular Ions from Volatile Samples. 364 A. Electron Ionization (EI) 364 B. Chemical Ionization (CI) 365 C. Photoionization (PI) 367 D. Field Ionization (FI) 367 IV. Molecular Ions from Nonvolatile Samples 367 Marvin L. Vestal received his B.S. and M.S. degrees, 1958 and 1960, A. Spray Techniques 367 respectively, in Engineering Sciences from Purdue Univesity, Layfayette, IN. In 1975 he received his Ph.D. degree in Chemical Physics from the B. Electrospray 367 University of Utah, Salt Lake City. From 1958 to 1960 he was a Scientist C. Desorption from Surfaces 369 at Johnston Laboratories, Inc., in Layfayette, IN. From 1960 to 1967 he D. High-Energy Particle Impact 369 became Senior Scientist at Johnston Laboratories, Inc., in Baltimore, MD. E. Low-Energy Particle Impact 370 From 1960 to 1962 he was a Graduate Student in the Department of Physics at John Hopkins University. From 1967 to 1970 he was Vice F. Low-Energy Impact with Liquid Surfaces 371 President at Scientific Research Instruments, Corp. in Baltimore, MD. From G. Flow FAB 371 1970 to 1975 he was a Graduate Student and Research Instructor at the H. Laser Ionization−MALDI 371 University of Utah, Salt Lake City. From 1976 to 1981 he became I. -

Selective Chemical Ionization of Nitrogen and Sulfur Heterocycles in Petroleum Fractions by Ion Trap Mass Spectrometry
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Selective Chemical Ionization of Nitrogen and Sulfur Heterocycles in Petroleum Fractions by Ion Trap Mass Spectrometry C. S. Creaser*, F. Krokos, and K. E. O’Neill Shod of Chemical Sciences, University of East Anglia, Norwich, United Kingdom M. J. C. Smith and P. G. McDowell BP Research, Sunbury-on-Thames, Middlesex, United Kingdom A procedure is reported for the selective ammonia chemical ionization of some nitrogen and sulfur heterocycles in petroleum fractions using ion trap mass spectrometry (ITMS). The ion trap scan routine is designed to optimize the population of ammonium reagent ions and eject from the trap (by radio frequency/direct current isolation) electron ionization products formed during reagent ion formation prior to ionization of the sample. The ITMS procedure is compared with standard ion trap detector and conventional quadrupole ammonia chemi- cal ionization for the determination of nitrogen and sulfur heterocycles in gas oil and kerosine samples. Greatly enhanced selectivity is shown for the ITMS procedure by sup res- sion of competing charge-exchange processes. (1 Am Sot Mass Spectrum 1993, 4, 322-326 ‘; he removal of organic compounds containing reagent ion NH;, is formed via the reactions [5] nitrogen and sulfur is an important part of the T petroleum-refining process 111. Their presence in NH, ‘1 NH;’ (1) petroleum distillates results in the formation of envi- NH, f NH:‘+ NH; + NH; (2) ronmental pollutants (SO,, NO,) during combustion, and their reactivity can lead to catalyst poisoning Whereas NH: is unreactive toward a wide range of in refinery processes. -

1 Introduction of Mass Spectrometry and Ambient Ionization Techniques
1 1 Introduction of Mass Spectrometry and Ambient Ionization Techniques Yiyang Dong, Jiahui Liu, and Tianyang Guo College of Life Science & Technology, Beijing University of Chemical Technology, No. 15 Beisanhuan East Road, Chaoyang District, Beijing, 100029, China 1.1 Evolution of Analytical Chemistry and Its Challenges in the Twenty-First Century The Chemical Revolution began in the eighteenth century, with the work of French chemist Antoine Lavoisier (1743–1794) representing a fundamental watershed that separated the “modern chemistry” era from the “protochemistry” era (Figure 1.1). However, analytical chemistry, a subdiscipline of chemistry, is an ancient science and its metrological tools, basic applications, and analytical processes can be dated back to early recorded history [1]. In chronological spans covering ancient times, the middle ages, the era of the nineteenth century, and the three chemical revolutionary periods, analytical chemistry has successfully evolved from the verge of the nineteenth century to modern and contemporary times, characterized by its versatile traits and unprecedented challenges in the twenty-first century. Historically, analytical chemistry can be termed as the mother of chemistry, as the nature and the composition of materials are always needed to be iden- tified first for specific utilizations subsequently; therefore, the development of analytical chemistry has always been ahead of general chemistry [2]. During pre-Hellenistic times when chemistry did not exist as a science, various ana- lytical processes, for example, qualitative touchstone method and quantitative fire-assay or cupellation scheme have been in existence as routine quality control measures for the purpose of noble goods authentication and anti-counterfeiting practices. Because of the unavailability of archeological clues for origin tracing, the chemical balance and the weights, as stated in the earliest documents ever found, was supposed to have been used only by the Gods [3]. -

Electrification Ionization: Fundamentals and Applications
Louisiana State University LSU Digital Commons LSU Doctoral Dissertations Graduate School 11-12-2019 Electrification Ionization: undamentalsF and Applications Bijay Kumar Banstola Louisiana State University and Agricultural and Mechanical College Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_dissertations Part of the Analytical Chemistry Commons Recommended Citation Banstola, Bijay Kumar, "Electrification Ionization: undamentalsF and Applications" (2019). LSU Doctoral Dissertations. 5103. https://digitalcommons.lsu.edu/gradschool_dissertations/5103 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Doctoral Dissertations by an authorized graduate school editor of LSU Digital Commons. For more information, please [email protected]. ELECTRIFICATION IONIZATION: FUNDAMENTALS AND APPLICATIONS A Dissertation Submitted to the Graduate Faculty of the Louisiana State University and Agricultural and Mechanical College in partial fulfillment of the requirements for the degree of Doctor of Philosophy in The Department of Chemistry by Bijay Kumar Banstola B. Sc., Northwestern State University of Louisiana, 2011 December 2019 This dissertation is dedicated to my parents: Tikaram and Shova Banstola my wife: Laxmi Kandel ii ACKNOWLEDGEMENTS I thank my advisor Professor Kermit K. Murray, for his continuous support and guidance throughout my Ph.D. program. Without his unwavering guidance and continuous help and encouragement, I could not have completed this program. I am also thankful to my committee members, Professor Isiah M. Warner, Professor Kenneth Lopata, and Professor Shengli Chen. I thank Dr. Fabrizio Donnarumma for his assistance and valuable insights to overcome the hurdles throughout my program. I appreciate Miss Connie David and Dr.