Electrification Ionization: Fundamentals and Applications
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mass Spectrometry: Quadrupole Mass Filter
Advanced Lab, Jan. 2008 Mass Spectrometry: Quadrupole Mass Filter The mass spectrometer is essentially an instrument which can be used to measure the mass, or more correctly the mass/charge ratio, of ionized atoms or other electrically charged particles. Mass spectrometers are now used in physics, geology, chemistry, biology and medicine to determine compositions, to measure isotopic ratios, for detecting leaks in vacuum systems, and in homeland security. Mass Spectrometer Designs The first mass spectrographs were invented almost 100 years ago, by A.J. Dempster, F.W. Aston and others, and have therefore been in continuous development over a very long period. However the principle of using electric and magnetic fields to accelerate and establish the trajectories of ions inside the spectrometer according to their mass/charge ratio is common to all the different designs. The following description of Dempster’s original mass spectrograph is a simple illustration of these physical principles: The magnetic sector spectrograph PUMP F DD S S3 1 r S2 Fig. 1: Dempster’s Mass Spectrograph (1918). Atoms/molecules are first ionized by electrons emitted from the hot filament (F) and then accelerated towards the entrance slit (S1). The ions then follow a semicircular trajectory established by the Lorentz force in a uniform magnetic field. The radius of the trajectory, r, is defined by three slits (S1, S2, and S3). Ions with this selected trajectory are then detected by the detector D. How the magnetic sector mass spectrograph works: Equating the Lorentz force with the centripetal force gives: qvB = mv2/r (1) where q is the charge on the ion (usually +e), B the magnetic field, m is the mass of the ion and r the radius of the ion trajectory. -

Gas Chromatography-Mass Spectroscopy
Gas Chromatography-Mass Spectroscopy Introduction Gas chromatography-mass spectroscopy (GC-MS) is one of the so-called hyphenated analytical techniques. As the name implies, it is actually two techniques that are combined to form a single method of analyzing mixtures of chemicals. Gas chromatography separates the components of a mixture and mass spectroscopy characterizes each of the components individually. By combining the two techniques, an analytical chemist can both qualitatively and quantitatively evaluate a solution containing a number of chemicals. Gas Chromatography In general, chromatography is used to separate mixtures of chemicals into individual components. Once isolated, the components can be evaluated individually. In all chromatography, separation occurs when the sample mixture is introduced (injected) into a mobile phase. In liquid chromatography (LC), the mobile phase is a solvent. In gas chromatography (GC), the mobile phase is an inert gas such as helium. The mobile phase carries the sample mixture through what is referred to as a stationary phase. The stationary phase is usually a chemical that can selectively attract components in a sample mixture. The stationary phase is usually contained in a tube of some sort called a column. Columns can be glass or stainless steel of various dimensions. The mixture of compounds in the mobile phase interacts with the stationary phase. Each compound in the mixture interacts at a different rate. Those that interact the fastest will exit (elute from) the column first. Those that interact slowest will exit the column last. By changing characteristics of the mobile phase and the stationary phase, different mixtures of chemicals can be separated. -

Qualitative and Quantitative Analysis
qualitative and quantitative analysis Russian scientist Tswett in 1906 used a glass columns packed with divided CaCO3(calcium carbonate) to separate plant pigments extracted by hexane. The pigments after separation appeared as colour bands that can come out of the column one by one. Tswett was the first to use the term "chromatography" derived from two Greek words "Chroma" meaning color and "graphein" meaning to write. Invention of Chromatography by M. Tswett Ether Chromatography Colors Chlorophyll CaCO3 5 *Definition of chromatography *Tswett (1906) stated that „chromatography is a method in which the components of a mixture are separated on adsorbent column in a flowing system”. *IUPAC definition (International Union of pure and applied Chemistry) (1993): Chromatography is a physical method of separation in which the components to be separated are distributed between two phases, one of which is stationary while the other moves in a definite direction. *Principles of Chromatography * Any chromatography system is composed of three components : * Stationary phase * Mobile phase * Mixture to be separated The separation process occurs because the components of mixture have different affinities for the two phases and thus move through the system at different rates. A component with a high affinity for the mobile phase moves quickly through the chromatographic system, whereas one with high affinity for the solid phase moves more slowly. *Forces Responsible for Separation * The affinity differences of the components for the stationary or the mobile phases can be due to several different chemical or physical properties including: * Ionization state * Polarity and polarizability * Hydrogen bonding / van der Waals’ forces * Hydrophobicity * Hydrophilicity * The rate at which a sample moves is determined by how much time it spends in the mobile phase. -

Theoretical Calculations for Electron Impact Ionization of Atoms and Molecules
Scholars' Mine Doctoral Dissertations Student Theses and Dissertations Fall 2017 Theoretical calculations for electron impact ionization of atoms and molecules Sadek Mohamed Fituri Amami Follow this and additional works at: https://scholarsmine.mst.edu/doctoral_dissertations Part of the Atomic, Molecular and Optical Physics Commons Department: Physics Recommended Citation Amami, Sadek Mohamed Fituri, "Theoretical calculations for electron impact ionization of atoms and molecules" (2017). Doctoral Dissertations. 2617. https://scholarsmine.mst.edu/doctoral_dissertations/2617 This thesis is brought to you by Scholars' Mine, a service of the Missouri S&T Library and Learning Resources. This work is protected by U. S. Copyright Law. Unauthorized use including reproduction for redistribution requires the permission of the copyright holder. For more information, please contact [email protected]. THEORETICAL CALCULATIONS FOR ELECTRON IMPACT IONIZATION OF ATOMS AND MOLECULES by SADEK MOHAMED FITURI AMAMI A DISSERTATION Presented to the Graduate Faculty of the MISSOURI UNIVERSITY OF SCIENCE AND TECHNOLOGY In Partial Fulfillment of the Requirements for the Degree DOCTOR OF PHILOSOPHY in PHYSICS 2017 Approved by Don H. Madison, Advisor Jerry L. Peacher Michael Schulz Daniel Fischer Gregory Gelles Copyright 2017 SADEK MOHAMED FITURI AMAMI All Rights Reserved iii PUBLICATION DISSERTATION OPTION This dissertation consists of the following ten articles which have been published as follows: Paper I: Pages 31-54 have been published by Phys Rev A.91.032707 (March 2015). Paper II: Pages 55-78 have been published by Phys Rev A.90.012704 (July 2014). Paper III: Pages 79-93 have been published by J. Phys. B: At. Mol. Opt. Phys. 49 185202 (September 2016). -

(ESI) Electrospray Ionization
Electrospray Ionization (ESI) • sample solution • voltage drop • solvent evaporates is injected between source, away as droplets directly into analyzer generates migrate across gap instrument ionized droplets “sheath gas” Electrospray Ionization (ESI) Injected solution usually includes ionizing agent H+ (from acid), Na+, K+ Large droplets from injected stream Resulting ions can be spontaneously eject singly or multiply smaller droplets (due to charged electrostatic repulsion) Electrospray Ionization (ESI) Multiply charged ions frequently observed. ESI M [M•H]+ + [M•2H]2+ + [M•3H]3+ + ... or [M•Na]+ + [M•2Na]2+ + [M•3Na]3+ + ... multiply lysozyme: charged big > 10 kDa protein (protonated) ions From this series of peaks, how do we determine mass of protein? Charge Ladders in Electrospray Ionization We know two things: 1. Any peak corresponds to [M•nH]n+; ion mass m =M+= M + n, charge z = n. 2. Peak to that peak’s left has one more proton attached, one more charge; ion mass m = M + n + 1, charge z = n + 1. Charge Ladders in Electrospray Ionization Set of simultaneous equations: (observed m/z #1) • z = M + z(H+) (observed m/z #2) • (z + 1) = M + z(H+) + 1 2 1 Charge Ladders in Electrospray Ionization Now performed by computer algorithm Set of simultaneous equations: that searches for ladders, outputs likely mass candidates. 1486 • z = M + z 1372.5 • (z + 1) = M + z + 1 Solution: z = 12 M = 17827.2 [M•13H]13+ 2 [M•12H]1 12+ actual MW lysozyme: 17825.2 If sample contains a mixture of counterions, can get messy. ESI-MS 22-mer single-stranded DNA (difficult to desalt) Atmospheric Pressure Chemical Ionization (APCI) •+ •+ Analogous to CI, but uses discharge to generate N2 /O2 , which then iiionizes evapora tdltllThthiited solvent molecules. -

Contents • Introduction to Proteomics and Mass Spectrometry
Contents • Introduction to Proteomics and Mass spectrometry - What we need to know in our quest to explain life - Fundamental things you need to know about mass spectrometry • Interfaces and ion sources - Electrospray ionization (ESI) - conventional and nanospray - Heated nebulizer atmospheric pressure chemical ionization - Matrix assisted laser desorption • Types of MS analyzers - Magnetic sector - Quadrupole - Time-of-flight - Hybrid - Ion trap In the next step in our quest to explain what is life The human genome project has largely been completed and many other genomes are surrendering to the gene sequencers. However, all this knowledge does not give us the information that is needed to explain how living cells work. To do that, we need to study proteins. In 2002, mass spectrometry has developed to the point where it has the capacity to obtain the "exact" molecular weight of many macromolecules. At the present time, this includes proteins up to 150,000 Da. Proteins of higher molecular weights (up to 500,000 Da) can also be studied by mass spectrometry, but with less accuracy. The paradigm for sequencing of peptides and identification of proteins has changed – because of the availability of the human genome database, peptides can be identified merely by their masses or by partial sequence information, often in minutes, not hours. This new capacity is shifting the emphasis of biomedical research back to the functional aspects of cell biochemistry, the expression of particular sets of genes and their gene products, the proteins of the cell. These are the new goals of the biological scientist: o to know which proteins are expressed in each cell, preferably one cell at a time o to know how these proteins are modified, information that cannot necessarily be deduced from the nucleotide sequence of individual genes. -

Laser Desorption/Ionization Time-Of-Flight Mass Spectrometry of Biomolecules Yu-Chen Chang Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1996 Laser desorption/ionization time-of-flight mass spectrometry of biomolecules Yu-chen Chang Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Analytical Chemistry Commons Recommended Citation Chang, Yu-chen, "Laser desorption/ionization time-of-flight mass spectrometry of biomolecules " (1996). Retrospective Theses and Dissertations. 11366. https://lib.dr.iastate.edu/rtd/11366 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. INFORMATION TO USERS This manuscript has been rqjroduced fix>m the microfihn master. UMI fihns the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, w^e others may be from any type of computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely afifect reproduction. In the unlikely event that the author did not send IMl a complete manuscript and there are misang pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. Oversize materials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand comer and continuing from left to right in equal sections with small overlaps. -

Separations Development and Application (WBS 2.4.1.101) U.S
Separations Development and Application (WBS 2.4.1.101) U.S. Department of Energy (DOE) Bioenergy Technologies Office (BETO) 2017 Project Peer Review James D. (“Jim”) McMillan NREL March 7, 2017 Biochemical Conversion Session This presentation does not contain any proprietary, confidential, or otherwise restricted information. Project Goal Overall goal: Define, develop and apply separation processes to enable cost- effective hydrocarbon fuel / precursor production; focus on sugars and fuel precursor streams; lipids pathway shown. Outcome: Down selected proven, viable methods for clarifying and concentrating the sugar intermediates stream and for recovering lipids from oleaginous yeast that pass “go” criteria (i.e., high yield, scalable, and cost effective). Relevance: Separations are key to overall process integration and economics; often represent ≥ 50% of total process costs; performance/efficiency can make or break process viability. Separations this project investigates – sugar stream clarification and concentration, and recovery of intracellular lipids from yeast – account for 17-26% of projected Minimum Fuel Selling Price (MFSP) for the integrated process. 2 Project Overview • Cost driven R&D to assess/develop/improve key process separations - Sugar stream separations: S/L, concentrative and polishing - Fuel precursor recovery separations: oleaginous yeast cell lysis and LLE lipid recovery • Identify and characterize effective methods - Show capability to pass relevant go/no-go criteria (e.g., high yield, low cost, scalable) • Exploit in situ separation for process intensification - Enable Continuous Enzymatic Hydrolysis (CEH) • Generate performance data to develop / refine process TEAs and LCAs 3 Separations Technoeconomic Impact $1.78 $2.18 $1.43 $0.79 4 Quad Chart Overview Timeline Barriers Primary focus on addressing upstream and Start: FY 15 (Oct., ‘14) downstream separations-related barriers: End: FY 17 (Sept., ‘17; projected) – Ct-G. -

Electron Ionization
Chapter 6 Chapter 6 Electron Ionization I. Introduction ......................................................................................................317 II. Ionization Process............................................................................................317 III. Strategy for Data Interpretation......................................................................321 1. Assumptions 2. The Ionization Process IV. Types of Fragmentation Pathways.................................................................328 1. Sigma-Bond Cleavage 2. Homolytic or Radical-Site-Driven Cleavage 3. Heterolytic or Charge-Site-Driven Cleavage 4. Rearrangements A. Hydrogen-Shift Rearrangements B. Hydride-Shift Rearrangements V. Representative Fragmentations (Spectra) of Classes of Compounds.......... 344 1. Hydrocarbons A. Saturated Hydrocarbons 1) Straight-Chain Hydrocarbons 2) Branched Hydrocarbons 3) Cyclic Hydrocarbons B. Unsaturated C. Aromatic 2. Alkyl Halides 3. Oxygen-Containing Compounds A. Aliphatic Alcohols B. Aliphatic Ethers C. Aromatic Alcohols D. Cyclic Ethers E. Ketones and Aldehydes F. Aliphatic Acids and Esters G. Aromatic Acids and Esters 4. Nitrogen-Containing Compounds A. Aliphatic Amines B. Aromatic Compounds Containing Atoms of Nitrogen C. Heterocyclic Nitrogen-Containing Compounds D. Nitro Compounds E. Concluding Remarks on the Mass Spectra of Nitrogen-Containing Compounds 5. Multiple Heteroatoms or Heteroatoms and a Double Bond 6. Trimethylsilyl Derivative 7. Determining the Location of Double Bonds VI. Library -

Modern Mass Spectrometry
Modern Mass Spectrometry MacMillan Group Meeting 2005 Sandra Lee Key References: E. Uggerud, S. Petrie, D. K. Bohme, F. Turecek, D. Schröder, H. Schwarz, D. Plattner, T. Wyttenbach, M. T. Bowers, P. B. Armentrout, S. A. Truger, T. Junker, G. Suizdak, Mark Brönstrup. Topics in Current Chemistry: Modern Mass Spectroscopy, pp. 1-302, 225. Springer-Verlag, Berlin, 2003. Current Topics in Organic Chemistry 2003, 15, 1503-1624 1 The Basics of Mass Spectroscopy ! Purpose Mass spectrometers use the difference in mass-to-charge ratio (m/z) of ionized atoms or molecules to separate them. Therefore, mass spectroscopy allows quantitation of atoms or molecules and provides structural information by the identification of distinctive fragmentation patterns. The general operation of a mass spectrometer is: "1. " create gas-phase ions "2. " separate the ions in space or time based on their mass-to-charge ratio "3. " measure the quantity of ions of each mass-to-charge ratio Ionization sources ! Instrumentation Chemical Ionisation (CI) Atmospheric Pressure CI!(APCI) Electron Impact!(EI) Electrospray Ionization!(ESI) SORTING DETECTION IONIZATION OF IONS OF IONS Fast Atom Bombardment (FAB) Field Desorption/Field Ionisation (FD/FI) Matrix Assisted Laser Desorption gaseous mass ion Ionisation!(MALDI) ion source analyzer transducer Thermospray Ionisation (TI) Analyzers quadrupoles vacuum signal Time-of-Flight (TOF) pump processor magnetic sectors 10-5– 10-8 torr Fourier transform and quadrupole ion traps inlet Detectors mass electron multiplier spectrum Faraday cup Ionization Sources: Classical Methods ! Electron Impact Ionization A beam of electrons passes through a gas-phase sample and collides with neutral analyte molcules (M) to produce a positively charged ion or a fragment ion. -
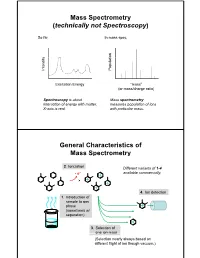
Mass Spectrometry (Technically Not Spectroscopy)
Mass Spectrometry (technically not Spectroscopy) So far, In mass spec, on y Populati Intensit Excitation Energy “mass” (or mass/charge ratio) Spectroscopy is about Mass spectrometry interaction of energy with matter. measures population of ions X-axis is real. with particular mass. General Characteristics of Mass Spectrometry 2. Ionization Different variants of 1-4 -e- available commercially. 4. Ion detection 1. Introduction of sample to gas phase (sometimes w/ separation) 3. Selection of one ion mass (Selection nearly always based on different flight of ion though vacuum.) General Components of a Mass Spectrometer Lots of choices, which can be mixed and matched. direct injection The Mass Spectrum fragment “daughter” ions M+ “parent” mass Sample Introduction: Direc t Inser tion Prob e If sample is a liquid, sample can also be injected directly into ionization region. If sample isn’t pure, get multiple parents (that can’t be distinguished from fragments). Capillary Column Introduction Continous source of molecules to spectrometer. detector column (including GC, LC, chiral, size exclusion) • Signal intensity depends on both amount of molecule and ionization efficiency • To use quantitatively, must calibrate peaks with respect eltilution time ttlitotal ion curren t to quantity eluted (TIC) over time Capillary Column Introduction Easy to interface with gas or liquid chromatography. TIC trace elution time time averaged time averaged mass spectrum mass spectrum Methods of Ionization: Electron Ionization (EI) 1 - + - 1 M + e (kV energy) M + 2e Fragmentation in Electron Ionization daughter ion (observed in spectrum) neutral fragment (not observed) excited parent at electron at electron energy of energy of 15 eV 70 e V Lower electron energy yields less fragmentation, but also less signal. -

Ion-To-Neutral Ratios and Thermal Proton Transfer in Matrix-Assisted Laser Desorption/Ionization
B American Society for Mass Spectrometry, 2015 J. Am. Soc. Mass Spectrom. (2015) 26:1242Y1251 DOI: 10.1007/s13361-015-1112-3 RESEARCH ARTICLE Ion-to-Neutral Ratios and Thermal Proton Transfer in Matrix-Assisted Laser Desorption/Ionization I-Chung Lu,1 Kuan Yu Chu,1,2 Chih-Yuan Lin,1 Shang-Yun Wu,1 Yuri A. Dyakov,1 Jien-Lian Chen,1 Angus Gray-Weale,3 Yuan-Tseh Lee,1,2 Chi-Kung Ni1,4 1Institute of Atomic and Molecular Sciences, Academia Sinica, Taipei, 10617, Taiwan 2Department of Chemistry, National Taiwan University, Taipei, 10617, Taiwan 3School of Chemistry, University of Melbourne, Melbourne, VIC 3010, Australia 4Department of Chemistry, National Tsing Hua University, Hsinchu, 30013, Taiwan Abstract. The ion-to-neutral ratios of four commonly used solid matrices, α-cyano-4- hydroxycinnamic acid (CHCA), 2,5-dihydroxybenzoic acid (2,5-DHB), sinapinic acid (SA), and ferulic acid (FA) in matrix-assisted laser desorption/ionization (MALDI) at 355 nm are reported. Ions are measured using a time-of-flight mass spectrometer combined with a time-sliced ion imaging detector. Neutrals are measured using a rotatable quadrupole mass spectrometer. The ion-to-neutral ratios of CHCA are three orders of magnitude larger than those of the other matrices at the same laser fluence. The ion-to-neutral ratios predicted using the thermal proton transfer model are similar to the experimental measurements, indicating that thermal proton transfer reactions play a major role in generating ions in ultraviolet-MALDI. Keywords: MALDI, Ionization mechanism, Thermal proton transfer, Ion-to-neutral ratio Received: 25 August 2014/Revised: 15 February 2015/Accepted: 16 February 2015/Published Online: 8 April 2015 Introduction in solid state UV-MALDI.