1 Introduction of Mass Spectrometry and Ambient Ionization Techniques
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Direct Analysis of Hops by Leaf Spray and Paper Spray Mass Spectrometry
Western Michigan University ScholarWorks at WMU Master's Theses Graduate College 12-2012 Direct Analysis of Hops by Leaf Spray and Paper Spray Mass Spectrometry Kari Blain Follow this and additional works at: https://scholarworks.wmich.edu/masters_theses Part of the Chemistry Commons Recommended Citation Blain, Kari, "Direct Analysis of Hops by Leaf Spray and Paper Spray Mass Spectrometry" (2012). Master's Theses. 100. https://scholarworks.wmich.edu/masters_theses/100 This Masters Thesis-Open Access is brought to you for free and open access by the Graduate College at ScholarWorks at WMU. It has been accepted for inclusion in Master's Theses by an authorized administrator of ScholarWorks at WMU. For more information, please contact [email protected]. DIRECT ANALYSIS OF HOPS BY LEAF SPRAY AND PAPER SPRAY MASS SPECTROMETRY Kari Blain, M.S. Western Michigan University, 2012 The objective of this research is to develop a new and innovative method of hops analysis, which is much faster than standard testing methods, as well as reduce the amount of consumables and solvent used. A detailed discussion on the development of an ambient ionization mass spectrometry method called paper spray (PS-MS) and leaf spray (LS-MS) mass spectrometry will be presented. This research investigates the use of PS-MS and LS-MS techniques to determine the α- and β- acids present in hops. PS-MS and LS-MS provide a fast way to analyze hops samples by delivering data as rapidly as a UV-Vis measurement while providing information similar to lengthy liquid chromatographic separations. The preliminary results shown here indicate that PS-MS could be used to determine cohumulone and α/β ratios. -

Chapter 8: Gravimetric Methods
Chapter 8 Gravimetric Methods Chapter Overview 8A Overview of Gravimetric Methods 8B Precipitation Gravimetry 8C Volatilization Gravimetry 8D Particulate Gravimetry 8E Key Terms 8F Chapter Summary 8G Problems 8H Solutions to Practice Exercises Gravimetry includes all analytical methods in which the analytical signal is a measurement of mass or a change in mass. When you step on a scale after exercising you are making, in a sense, a gravimetric determination of your mass. Mass is the most fundamental of all analytical measurements, and gravimetry is unquestionably our oldest quantitative analytical technique. The publication in 1540 of Vannoccio Biringuccio’sPirotechnia is an early example of applying gravimetry—although not yet known by this name—to the analysis of metals and ores.1 Although gravimetry no longer is the most important analytical method, it continues to find use in specialized applications. 1 Smith, C. S.; Gnodi, M. T. translation of Biringuccio, V. Pirotechnia, MIT Press: Cambridge, MA, 1959. 355 356 Analytical Chemistry 2.0 8A Overview of Gravimetric Methods Before we consider specific gravimetric methods, let’s take a moment to develop a broad survey of gravimetry. Later, as you read through the de- scriptions of specific gravimetric methods, this survey will help you focus on their similarities instead of their differences. You will find that it is easier to understand a new analytical method when you can see its relationship to other similar methods. 8A.1 Using Mass as an Analytical Signal Method 2540D in Standard Methods for Suppose you are to determine the total suspended solids in the water re- the Examination of Waters and Wastewaters, leased by a sewage-treatment facility. -

Multiplatform Investigation of Plasma and Tissue Lipid Signatures of Breast Cancer Using Mass Spectrometry Tools
International Journal of Molecular Sciences Article Multiplatform Investigation of Plasma and Tissue Lipid Signatures of Breast Cancer Using Mass Spectrometry Tools 1, 2, 2 3 Alex Ap. Rosini Silva y, Marcella R. Cardoso y , Luciana Montes Rezende , John Q. Lin , Fernando Guimaraes 2 , Geisilene R. Paiva Silva 4, Michael Murgu 5, Denise Gonçalves Priolli 1, Marcos N. Eberlin 6, Alessandra Tata 7, Livia S. Eberlin 3 , Sophie F. M. Derchain 2 and Andreia M. Porcari 1,* 1 Postgraduate Program of Health Sciences, São Francisco University, Bragança Paulista SP 12916-900, Brazil; [email protected] (A.A.R.S.); [email protected] (D.G.P.) 2 Department of Gynecological and Breast Oncology, Women’s Hospital (CAISM), Faculty of Medical Sciences, State University of Campinas (UNICAMP), Campinas SP 13083-881, Brazil; [email protected] (M.R.C.); [email protected] (L.M.R.); [email protected] (F.G.); [email protected] (S.F.M.D.) 3 Department of Chemistry, The University of Texas at Austin, Austin, TX 78712, USA; [email protected] (J.Q.L.); [email protected] (L.S.E.) 4 Laboratory of Molecular and Investigative Pathology—LAPE, Women’s Hospital (CAISM), Faculty of Medical Sciences, State University of Campinas (UNICAMP), Campinas SP 13083-881, Brazil; [email protected] 5 Waters Corporation, São Paulo, SP 13083-970, Brazil; [email protected] 6 School of Engineering, Mackenzie Presbyterian University, São Paulo SP 01302-907, Brazil; [email protected] 7 Laboratorio di Chimica Sperimentale, Istituto Zooprofilattico Sperimentale delle Venezie, Viale Fiume 78, 36100 Vicenza, Italy; [email protected] * Correspondence: [email protected]; Tel.: +55-11-2454-8047 These authors contributed equally to this work. -

Theoretical Calculations for Electron Impact Ionization of Atoms and Molecules
Scholars' Mine Doctoral Dissertations Student Theses and Dissertations Fall 2017 Theoretical calculations for electron impact ionization of atoms and molecules Sadek Mohamed Fituri Amami Follow this and additional works at: https://scholarsmine.mst.edu/doctoral_dissertations Part of the Atomic, Molecular and Optical Physics Commons Department: Physics Recommended Citation Amami, Sadek Mohamed Fituri, "Theoretical calculations for electron impact ionization of atoms and molecules" (2017). Doctoral Dissertations. 2617. https://scholarsmine.mst.edu/doctoral_dissertations/2617 This thesis is brought to you by Scholars' Mine, a service of the Missouri S&T Library and Learning Resources. This work is protected by U. S. Copyright Law. Unauthorized use including reproduction for redistribution requires the permission of the copyright holder. For more information, please contact [email protected]. THEORETICAL CALCULATIONS FOR ELECTRON IMPACT IONIZATION OF ATOMS AND MOLECULES by SADEK MOHAMED FITURI AMAMI A DISSERTATION Presented to the Graduate Faculty of the MISSOURI UNIVERSITY OF SCIENCE AND TECHNOLOGY In Partial Fulfillment of the Requirements for the Degree DOCTOR OF PHILOSOPHY in PHYSICS 2017 Approved by Don H. Madison, Advisor Jerry L. Peacher Michael Schulz Daniel Fischer Gregory Gelles Copyright 2017 SADEK MOHAMED FITURI AMAMI All Rights Reserved iii PUBLICATION DISSERTATION OPTION This dissertation consists of the following ten articles which have been published as follows: Paper I: Pages 31-54 have been published by Phys Rev A.91.032707 (March 2015). Paper II: Pages 55-78 have been published by Phys Rev A.90.012704 (July 2014). Paper III: Pages 79-93 have been published by J. Phys. B: At. Mol. Opt. Phys. 49 185202 (September 2016). -

Gravimetric Analysis
Gravimetric Analysis Gravimetric Analysis can be used to determine the percent composition of a species in a compound. This is known as quantitative analysis. An agent (precipitating agent) is chosen which will cause the species of interest to precipitate out of solution. The precipitate is then isolated and weighed. Through a series of calculations, the amount of the species in the original sample or compound can then be determined. 1. Weigh accurately about 0.5 g of the unknown, and dissolve it in water. The volume of water is not important, but you do want to make sure the solid is completely dissolved. 2. Add the precipitating agent in the form of solution. For example, to precipitate a chloride use a soluble salt of silver (AgNO3). 3. Add the precipitating agent until no more solid seems to precipitate. Allow the mixture to sit for about 5 minutes, and then add a few drops more, observing to see if any more precipitation occurs. If you see more solid forming, keep repeating the steps until no more precipitate results. Now separate the solid from the liquid. The mixture can be filtered by vacuum filtration through a weighed filter paper. Other methods involve gravity filtration or use of a centrifuge. Allow the solid to dry until you get a constant weight for readings at subsequent times (at least overnight or through use of an oven). 4. From the mass of precipitated compound (ppt) and the formula (in this case AgCl), you can calculate the percent composition of a species (in this case Cl-) in the original sample. -

Electron Ionization
Chapter 6 Chapter 6 Electron Ionization I. Introduction ......................................................................................................317 II. Ionization Process............................................................................................317 III. Strategy for Data Interpretation......................................................................321 1. Assumptions 2. The Ionization Process IV. Types of Fragmentation Pathways.................................................................328 1. Sigma-Bond Cleavage 2. Homolytic or Radical-Site-Driven Cleavage 3. Heterolytic or Charge-Site-Driven Cleavage 4. Rearrangements A. Hydrogen-Shift Rearrangements B. Hydride-Shift Rearrangements V. Representative Fragmentations (Spectra) of Classes of Compounds.......... 344 1. Hydrocarbons A. Saturated Hydrocarbons 1) Straight-Chain Hydrocarbons 2) Branched Hydrocarbons 3) Cyclic Hydrocarbons B. Unsaturated C. Aromatic 2. Alkyl Halides 3. Oxygen-Containing Compounds A. Aliphatic Alcohols B. Aliphatic Ethers C. Aromatic Alcohols D. Cyclic Ethers E. Ketones and Aldehydes F. Aliphatic Acids and Esters G. Aromatic Acids and Esters 4. Nitrogen-Containing Compounds A. Aliphatic Amines B. Aromatic Compounds Containing Atoms of Nitrogen C. Heterocyclic Nitrogen-Containing Compounds D. Nitro Compounds E. Concluding Remarks on the Mass Spectra of Nitrogen-Containing Compounds 5. Multiple Heteroatoms or Heteroatoms and a Double Bond 6. Trimethylsilyl Derivative 7. Determining the Location of Double Bonds VI. Library -

Modern Mass Spectrometry
Modern Mass Spectrometry MacMillan Group Meeting 2005 Sandra Lee Key References: E. Uggerud, S. Petrie, D. K. Bohme, F. Turecek, D. Schröder, H. Schwarz, D. Plattner, T. Wyttenbach, M. T. Bowers, P. B. Armentrout, S. A. Truger, T. Junker, G. Suizdak, Mark Brönstrup. Topics in Current Chemistry: Modern Mass Spectroscopy, pp. 1-302, 225. Springer-Verlag, Berlin, 2003. Current Topics in Organic Chemistry 2003, 15, 1503-1624 1 The Basics of Mass Spectroscopy ! Purpose Mass spectrometers use the difference in mass-to-charge ratio (m/z) of ionized atoms or molecules to separate them. Therefore, mass spectroscopy allows quantitation of atoms or molecules and provides structural information by the identification of distinctive fragmentation patterns. The general operation of a mass spectrometer is: "1. " create gas-phase ions "2. " separate the ions in space or time based on their mass-to-charge ratio "3. " measure the quantity of ions of each mass-to-charge ratio Ionization sources ! Instrumentation Chemical Ionisation (CI) Atmospheric Pressure CI!(APCI) Electron Impact!(EI) Electrospray Ionization!(ESI) SORTING DETECTION IONIZATION OF IONS OF IONS Fast Atom Bombardment (FAB) Field Desorption/Field Ionisation (FD/FI) Matrix Assisted Laser Desorption gaseous mass ion Ionisation!(MALDI) ion source analyzer transducer Thermospray Ionisation (TI) Analyzers quadrupoles vacuum signal Time-of-Flight (TOF) pump processor magnetic sectors 10-5– 10-8 torr Fourier transform and quadrupole ion traps inlet Detectors mass electron multiplier spectrum Faraday cup Ionization Sources: Classical Methods ! Electron Impact Ionization A beam of electrons passes through a gas-phase sample and collides with neutral analyte molcules (M) to produce a positively charged ion or a fragment ion. -
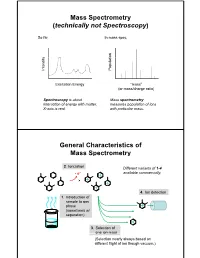
Mass Spectrometry (Technically Not Spectroscopy)
Mass Spectrometry (technically not Spectroscopy) So far, In mass spec, on y Populati Intensit Excitation Energy “mass” (or mass/charge ratio) Spectroscopy is about Mass spectrometry interaction of energy with matter. measures population of ions X-axis is real. with particular mass. General Characteristics of Mass Spectrometry 2. Ionization Different variants of 1-4 -e- available commercially. 4. Ion detection 1. Introduction of sample to gas phase (sometimes w/ separation) 3. Selection of one ion mass (Selection nearly always based on different flight of ion though vacuum.) General Components of a Mass Spectrometer Lots of choices, which can be mixed and matched. direct injection The Mass Spectrum fragment “daughter” ions M+ “parent” mass Sample Introduction: Direc t Inser tion Prob e If sample is a liquid, sample can also be injected directly into ionization region. If sample isn’t pure, get multiple parents (that can’t be distinguished from fragments). Capillary Column Introduction Continous source of molecules to spectrometer. detector column (including GC, LC, chiral, size exclusion) • Signal intensity depends on both amount of molecule and ionization efficiency • To use quantitatively, must calibrate peaks with respect eltilution time ttlitotal ion curren t to quantity eluted (TIC) over time Capillary Column Introduction Easy to interface with gas or liquid chromatography. TIC trace elution time time averaged time averaged mass spectrum mass spectrum Methods of Ionization: Electron Ionization (EI) 1 - + - 1 M + e (kV energy) M + 2e Fragmentation in Electron Ionization daughter ion (observed in spectrum) neutral fragment (not observed) excited parent at electron at electron energy of energy of 15 eV 70 e V Lower electron energy yields less fragmentation, but also less signal. -

Portable Mass Spectrometry
Food Analytical Methods https://doi.org/10.1007/s12161-019-01666-6 Potential of Recent Ambient Ionization Techniques for Future Food Contaminant Analysis Using (Trans)Portable Mass Spectrometry Marco H. Blokland1 & Arjen Gerssen1 & Paul W. Zoontjes1 & Janusz Pawliszyn2 & Michel W.F. Nielen1,3 Received: 3 June 2019 /Accepted: 31 October 2019 # The Author(s) 2019 Abstract In food analysis, a trend towards on-site testing of quality and safety parameters is emerging. So far, on-site testing has been mainly explored by miniaturized optical spectroscopy and ligand-binding assay approaches such as lateral flow immunoassays and biosensors. However, for the analysis of multiple parameters at regulatory levels, mass spectrometry (MS) is the method of choice in food testing laboratories. Thanks to recent developments in ambient ionization and upcoming miniaturization of mass analyzers, (trans)portable mass spectrometry may be added to the toolkit for on-site testing and eventually compete with multiplex immunoassays in mixture analysis. In this study, we preliminary evaluated a selection of recent ambient ionization techniques for their potential in simplified testing of selected food contaminants such as pesticides, veterinary drugs, and natural toxins, aiming for a minimum in sample preparation while maintaining acceptable sensitivity and robustness. Matrix-assisted inlet ionization (MAI), handheld desorption atmospheric pressure chemical ionization (DAPCI), transmission-mode direct analysis in real time (TM-DART), and coated blade spray (CBS) were coupled to both benchtop Orbitrap and compact quad- rupole single-stage mass analyzers, while CBS was also briefly studied on a benchtop triple-quadrupole MS. From the results, it can be concluded that for solid and liquid sample transmission configurations provide the highest sensitivity while upon addition ofastationaryphase,suchasinCBS,evenlowμg/L levels in urine samples can be achieved provided the additional selectivity of tandem mass spectrometry is exploited. -

Advances in Ionization for Mass Spectrometry † ‡ ‡ Patricia M
Review pubs.acs.org/ac Advances in Ionization for Mass Spectrometry † ‡ ‡ Patricia M. Peacock, Wen-Jing Zhang, and Sarah Trimpin*, † First State IR, LLC, 118 Susan Drive, Hockessin, Delaware 19707, United States ‡ Department of Chemistry, Wayne State University, 5101 Cass Avenue, Detroit, Michigan 48202, United States ■ CONTENTS ■ EMERGING DEVELOPMENTS RELATED TO NEWER IONIZATION TECHNOLOGIES Scope 372 Emerging Developments Related to Newer Ioniza- A relatively new means of transferring small, large, volatile, and tion Technologies 372 nonvolatile compounds from the solid or solution state directly Electrospray-Based Ambient Ionization 375 into the gas phase as ions, falling under the general term inlet Discharge-Based Ambient Ionization 377 ionization and individually termed matrix-assisted ionization Traditional Ionization Methods for MS 381 (MAI), solvent-assisted ionization (SAI), or laserspray Technologies Using a Laser for Ionization 381 ionization inlet (LSI) if a laser is used, continues to be API Ionization 383 developed as a sensitive and robust ionization method in MS. Author Information 384 In MAI, specific small molecule matrix compounds have been Corresponding Author 384 discovered which require only exposure to subatmospheric ORCID 384 pressure conditions, by default available with all mass Notes 384 spectrometers, to produce gas-phase ions of peptides, proteins, Biographies 384 lipids, synthetic polymers, and carbohydrates for analysis by Acknowledgments 385 MS.16 Some MAI matrixes produce abundant ions without the References 385 need to apply thermal energy. Out of over 40 such matrixes, the top 10 have been categorized for positive and negative mode measurements. These matrixes sublime when placed under subatmospheric pressure so that, similar to solvents, they are pumped from the mass spectrometer eliminating matrix contamination.17 Several studies related to MAI were published in this period ■ SCOPE by Trimpin and collaborators. -

An Organic Chemist's Guide to Electrospray Mass Spectrometric
molecules Review An Organic Chemist’s Guide to Electrospray Mass Spectrometric Structure Elucidation Arnold Steckel 1 and Gitta Schlosser 2,* 1 Hevesy György PhD School of Chemistry, ELTE Eötvös Loránd University, Pázmány Péter sétány 1/A, 1117 Budapest, Hungary; [email protected] 2 Department of Analytical Chemistry, ELTE Eötvös Loránd University, Pázmány Péter sétány 1/A, 1117 Budapest, Hungary * Correspondence: [email protected] Received: 16 January 2019; Accepted: 8 February 2019; Published: 10 February 2019 Abstract: Tandem mass spectrometry is an important tool for structure elucidation of natural and synthetic organic products. Fragmentation of odd electron ions (OE+) generated by electron ionization (EI) was extensively studied in the last few decades, however there are only a few systematic reviews available concerning the fragmentation of even-electron ions (EE+/EE−) produced by the currently most common ionization techniques, electrospray ionization (ESI) and atmospheric pressure chemical ionization (APCI). This review summarizes the most important features of tandem mass spectra generated by collision-induced dissociation fragmentation and presents didactic examples for the unexperienced users. Keywords: tandem mass spectrometry; MS/MS fragmentation; collision-induced dissociation; CID; ESI; structure elucidation 1. Introduction Electron ionization (EI), a hard ionization technique, is the method of choice for analyses of small (<1000 Da), nonpolar, volatile compounds. As its name implies, the technique involves ionization by electrons with ~70 eV energy. This energy is high enough to yield very reproducible mass spectra with a large number of fragments. However, these spectra frequently lack the radical type molecular ions (M+) due to the high internal energy transferred to the precursors [1]. -

Lecture 10. Analytical Chemistry What Is Analytical Chemistry ?
5/5/2019 What is Analytical Chemistry ? It deals with: Lecture 10. • separation Analytical Chemistry • identification • determination of components in a sample. Basic concepts It includes coverage of chemical equilibrium and statistical treatment of data. It encompasses any type of tests that provide information relating to the chemical composition of a sample. 1 2 • Analytical chemistry is divided into two areas of analysis: • The substance to be analyzed within a sample • Qualitative – recognizes the particles which are present in a sample. is known as an analyte, whereas the substances which may cause incorrect or • Quantitative – identifies how much of inaccurate results are known as chemical particles is present in a sample. interferents. 3 4 1 5/5/2019 Qualitative analysis is used to separate an analyte from interferents existing in a sample and detect the previous one. It gives negative, positive, or yes/no types of data. Qualitative analysis It informs whether or not the analyte is present in a sample. 5 6 Examples of qualitative analysis 7 8 2 5/5/2019 Analysis of an inorganic sample The classical procedure for systematic analysis of an inorganic sample consists of several parts: preliminary tests (heating, solubility in water, appearance of moisture) more complicated tests e.g. introducing the sample into a flame and noting the colour produced; determination of anionic or cationic constituents of 9 solute dissolved in water 10 Flame test Sodium Bright yellow (intense, persistent) Potassium Pale violet (slight, fleeting) Solutions of ions, when mixed with concentrated Calcium Brick red (medium, fleeting) HCl and heated on a nickel/chromium wire in a flame, cause the flame to change to a colour Strontium Crimson (medium) characteristic of the element.