Gas Chromatography-Mass Spectroscopy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Electrochemical Real-Time Mass Spectrometry: a Novel Tool for Time-Resolved Characterization of the Products of Electrochemical Reactions
Electrochemical real-time mass spectrometry: A novel tool for time-resolved characterization of the products of electrochemical reactions Elektrochemische Realzeit-Massenspektrometrie: Eine neuartige Methode zur zeitaufgelösten Charakterisierung der Produkte elektrochemischer Reaktionen Der Technischen Fakultät der Friedrich-Alexander-Universität Erlangen-Nürnberg zur Erlangung des Doktorgrades Dr.-Ingenieur vorgelegt von Peyman Khanipour Mehrin aus Shiraz, Iran Als Dissertation genehmigt von der Technischen Fakultät der Friedrich-Alexander-Universität Erlangen-Nürnberg Tag der mündlichen Prüfung: 17.11.2020 Vorsitzender des Promotionsorgans: Prof. Dr.-Ing. habil. Andreas Paul Fröba Gutachter: Prof. Dr. Karl J.J. Mayrhofer Prof. Dr. Frank-Michael Matysik I Acknowledgements This study is done in the electrosynthesis team of the electrocatalysis unit at Helmholtz- Institut Erlangen-Nürnberg (HI ERN) with the financial support of Forschungszentrum Jülich. I would like to express my deep gratitude to Prof. Dr. Karl J. J. Mayrhofer for accepting me as a Ph.D. student and also for all his encouragement, supports, and freedoms during my study. I’m grateful to Prof. Dr. Frank-Michael Matysik for kindly accepting to act as a second reviewer and also for the time he has invested in reading this thesis. This piece of work is enabled by collaboration with scientists from different expertise. I would like to express my appreciation to Dr. Sandra Haschke from FAU for providing shape-controlled high surface area platinum electrodes which I used for performing oxidation of primary alcohols and also the characterization of the provided material SEM, EDX, and XRD. Mr. Mario Löffler from HI ERN for obtaining the XPS data and his remarkable knowledge with the interpretation of the spectra on copper-based electrodes for the CO 2 electroreduction reaction. -

22 Chromatography and Mass Spectrometer
MODULE Chromatography and Mass Spectrometer Biochemistry 22 Notes CHROMATOGRAPHY AND MASS SPECTROMETER 22.1 INTRODUCTION We know that the biochemistry or biological chemistry deals with the study of molecules present in organisms. These molecules are called as biomolecules and they form the basic unit of every cell. These include carbohydrates, proteins, lipids and nucleic acids. To study the biomolecules and to know their function, they have to be obtained in purified form. Purification of the biomolecules includes many physical and chemical methods. This topic gives about two of the commonly used methods namely, chromatography and mass spectrometry. These methods deals with purification and separation of biomolecules namely, protein and nucleic acids. OBJECTIVES After reading this lesson, you will be able to: z define the chromatography and mass spectrometry z describe the principle and important types of chromatographic methods z describe the principle and components of a mass spectrometer z enlist types of mass spectrometer z describe various uses of mass spectrometry 22.2 CHROMATOGRAPHY When we have a mixture of colored small beads, it is easily separated by visual examination. The same holds true for many chemical molecules. In 1903, 280 BIOCHEMISTRY Chromatography and Mass Spectrometer MODULE Mikhail, a botanist (person studies plants) described the separation of leaf Biochemistry pigments (different colors) in solution by using solid adsorbents. He named this method of separation called chromatography. It comes from two Greek words: chroma – colour graphein – to write/detect Modern separation methods are based on different types of chromatographic methods. The basic principle of any chromatography is due to presence of two Notes phases: z Mobile phase – substances to be separated are mixed with this fluid; it may be gas or liquid; it continues moves through the chromatographic instrument z Stationary phase – it does not move; it is packed inside a column; it is a porous matrix that helps in separation of substances present in sample. -

Qualitative and Quantitative Analysis
qualitative and quantitative analysis Russian scientist Tswett in 1906 used a glass columns packed with divided CaCO3(calcium carbonate) to separate plant pigments extracted by hexane. The pigments after separation appeared as colour bands that can come out of the column one by one. Tswett was the first to use the term "chromatography" derived from two Greek words "Chroma" meaning color and "graphein" meaning to write. Invention of Chromatography by M. Tswett Ether Chromatography Colors Chlorophyll CaCO3 5 *Definition of chromatography *Tswett (1906) stated that „chromatography is a method in which the components of a mixture are separated on adsorbent column in a flowing system”. *IUPAC definition (International Union of pure and applied Chemistry) (1993): Chromatography is a physical method of separation in which the components to be separated are distributed between two phases, one of which is stationary while the other moves in a definite direction. *Principles of Chromatography * Any chromatography system is composed of three components : * Stationary phase * Mobile phase * Mixture to be separated The separation process occurs because the components of mixture have different affinities for the two phases and thus move through the system at different rates. A component with a high affinity for the mobile phase moves quickly through the chromatographic system, whereas one with high affinity for the solid phase moves more slowly. *Forces Responsible for Separation * The affinity differences of the components for the stationary or the mobile phases can be due to several different chemical or physical properties including: * Ionization state * Polarity and polarizability * Hydrogen bonding / van der Waals’ forces * Hydrophobicity * Hydrophilicity * The rate at which a sample moves is determined by how much time it spends in the mobile phase. -

Ultra-Performance Liquid Chromatography Coupled to Quadrupole-Orthogonal Time-Of-flight Mass Spectrometry
RAPID COMMUNICATIONS IN MASS SPECTROMETRY Rapid Commun. Mass Spectrom. 2004; 18: 2331–2337 Published online in Wiley InterScience (www.interscience.wiley.com). DOI: 10.1002/rcm.1627 Ultra-performance liquid chromatography coupled to quadrupole-orthogonal time-of-flight mass spectrometry Robert Plumb1*, Jose Castro-Perez2, Jennifer Granger1, Iain Beattie3, Karine Joncour3 and Andrew Wright3 1Waters Corporation, Milford, MA, USA 2Waters Corporation, MS Technology Center, Manchester, UK 3AstraZeneca R&D Charnwood, Physical & Metabolic Science, Loughborough, UK Received 19 June 2004; Revised 5 August 2004; Accepted 9 August 2004 Ultra-performance liquid chromatography (UPLC) utilizes sub-2 mm particles with high linear sol- vent velocities to effect dramatic increases in resolution, sensitivity and speed of analysis. The reduction in particle size to below 2 mm requires instrumentation that can operate at pressures in the 6000–15 000 psi range. The typical peak widths generated by the UPLC system are in the order of 1–2 s for a 10-min separation. In the present work this technology has been applied to the study of in vivo drug metabolism, in particular the analysis of drug metabolites in bile. The reduction in peak width significantly increases analytical sensitivity by three- to five-fold, and the reduction in peak width, and concomitant increase in peak capacity, significantly reduces spectral overlap resulting in superior spectral quality in both MS and MS/MS modes. The application of UPLC/ MS resulted in the detection of additional drug metabolites, superior separation and improved spectral quality. Copyright # 2004 John Wiley & Sons, Ltd. The detection and identification of drug metabolites is crucial Liquid chromatography/mass spectrometry (LC/MS) and to both the drug discovery and development processes, LC/MS/MS have become the mainstay of the drug metabo- although in these two areas the emphasis is slightly different. -

Coupling Gas Chromatography to Mass Spectrometry
Coupling Gas Chromatography to Mass Spectrometry Introduction The suite of gas chromatographic detectors includes (roughly in order from most common to the least): the flame ionization detector (FID), thermal conductivity detector (TCD or hot wire detector), electron capture detector (ECD), photoionization detector (PID), flame photometric detector (FPD), thermionic detector, and a few more unusual or VERY expensive choices like the atomic emission detector (AED) and the ozone- or fluorine-induce chemiluminescence detectors. All of these except the AED produce an electrical signal that varies with the amount of analyte exiting the chromatographic column. The AED does that AND yields the emission spectrum of selected elements in the analytes as well. Another GC detector that is also very expensive but very powerful is a scaled down version of the mass spectrometer. When coupled to a GC the detection system itself is often referred to as the mass selective detector or more simply the mass detector. This powerful analytical technique belongs to the class of hyphenated analytical instrumentation (since each part had a different beginning and can exist independently) and is called gas chromatograhy/mass spectrometry (GC/MS). Placed at the end of a capillary column in a manner similar to the other GC detectors, the mass detector is more complicated than, for instance, the FID because of the mass spectrometer's complex requirements for the process of creation, separation, and detection of gas phase ions. A capillary column is required in the chromatograph because the entire MS process must be carried out at very low pressures (~10-5 torr) and in order to meet this requirement a vacuum is maintained via constant pumping using a vacuum pump. -

Laser Desorption/Ionization Time-Of-Flight Mass Spectrometry of Biomolecules Yu-Chen Chang Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1996 Laser desorption/ionization time-of-flight mass spectrometry of biomolecules Yu-chen Chang Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Analytical Chemistry Commons Recommended Citation Chang, Yu-chen, "Laser desorption/ionization time-of-flight mass spectrometry of biomolecules " (1996). Retrospective Theses and Dissertations. 11366. https://lib.dr.iastate.edu/rtd/11366 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. INFORMATION TO USERS This manuscript has been rqjroduced fix>m the microfihn master. UMI fihns the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, w^e others may be from any type of computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely afifect reproduction. In the unlikely event that the author did not send IMl a complete manuscript and there are misang pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. Oversize materials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand comer and continuing from left to right in equal sections with small overlaps. -

Separations Development and Application (WBS 2.4.1.101) U.S
Separations Development and Application (WBS 2.4.1.101) U.S. Department of Energy (DOE) Bioenergy Technologies Office (BETO) 2017 Project Peer Review James D. (“Jim”) McMillan NREL March 7, 2017 Biochemical Conversion Session This presentation does not contain any proprietary, confidential, or otherwise restricted information. Project Goal Overall goal: Define, develop and apply separation processes to enable cost- effective hydrocarbon fuel / precursor production; focus on sugars and fuel precursor streams; lipids pathway shown. Outcome: Down selected proven, viable methods for clarifying and concentrating the sugar intermediates stream and for recovering lipids from oleaginous yeast that pass “go” criteria (i.e., high yield, scalable, and cost effective). Relevance: Separations are key to overall process integration and economics; often represent ≥ 50% of total process costs; performance/efficiency can make or break process viability. Separations this project investigates – sugar stream clarification and concentration, and recovery of intracellular lipids from yeast – account for 17-26% of projected Minimum Fuel Selling Price (MFSP) for the integrated process. 2 Project Overview • Cost driven R&D to assess/develop/improve key process separations - Sugar stream separations: S/L, concentrative and polishing - Fuel precursor recovery separations: oleaginous yeast cell lysis and LLE lipid recovery • Identify and characterize effective methods - Show capability to pass relevant go/no-go criteria (e.g., high yield, low cost, scalable) • Exploit in situ separation for process intensification - Enable Continuous Enzymatic Hydrolysis (CEH) • Generate performance data to develop / refine process TEAs and LCAs 3 Separations Technoeconomic Impact $1.78 $2.18 $1.43 $0.79 4 Quad Chart Overview Timeline Barriers Primary focus on addressing upstream and Start: FY 15 (Oct., ‘14) downstream separations-related barriers: End: FY 17 (Sept., ‘17; projected) – Ct-G. -

Electron Ionization
Chapter 6 Chapter 6 Electron Ionization I. Introduction ......................................................................................................317 II. Ionization Process............................................................................................317 III. Strategy for Data Interpretation......................................................................321 1. Assumptions 2. The Ionization Process IV. Types of Fragmentation Pathways.................................................................328 1. Sigma-Bond Cleavage 2. Homolytic or Radical-Site-Driven Cleavage 3. Heterolytic or Charge-Site-Driven Cleavage 4. Rearrangements A. Hydrogen-Shift Rearrangements B. Hydride-Shift Rearrangements V. Representative Fragmentations (Spectra) of Classes of Compounds.......... 344 1. Hydrocarbons A. Saturated Hydrocarbons 1) Straight-Chain Hydrocarbons 2) Branched Hydrocarbons 3) Cyclic Hydrocarbons B. Unsaturated C. Aromatic 2. Alkyl Halides 3. Oxygen-Containing Compounds A. Aliphatic Alcohols B. Aliphatic Ethers C. Aromatic Alcohols D. Cyclic Ethers E. Ketones and Aldehydes F. Aliphatic Acids and Esters G. Aromatic Acids and Esters 4. Nitrogen-Containing Compounds A. Aliphatic Amines B. Aromatic Compounds Containing Atoms of Nitrogen C. Heterocyclic Nitrogen-Containing Compounds D. Nitro Compounds E. Concluding Remarks on the Mass Spectra of Nitrogen-Containing Compounds 5. Multiple Heteroatoms or Heteroatoms and a Double Bond 6. Trimethylsilyl Derivative 7. Determining the Location of Double Bonds VI. Library -

Rubber-Modified Epoxies: in Situ Detection of the Phase Separation by Differential Scanning Calorimetry
Rubber-modified epoxies: in situ detection of the phase separation by differential scanning calorimetry Dong Pu Fang*, C. C. Riccardi and R. J. J. Williamst Institute of Materials Science and Technology (INTEMA), University of Mar de/Plata and National Research Council (CONICET), J. B. Justo 4302, 7600 Mar de/Plata, Argentina (Received 18 May 1993) The phase separation of a rubber in the course of a thermosetting polymerization is associated with heat release. Mixing is endothermic as revealed by the positive value of the interaction parameter while demixing is exothermic. Differential scanning calorimetry may be used to detect heat effects related to phase separation. Experimental results are reported for an unreactive system (castor oil/epoxy resin) and reactive systems (modifier/epoxy resin/diamine) using either castor oil or epoxy-terminated copolymers of acrylonitrile and butadiene as modifiers. (Keywords: rubber-modified epoxies; phase separation; d.s.c.) Introduction represents the residual free energy of mixing (part or all A common method to toughen epoxy resins is to of it may be considered as an enthalpic contribution). dissolve rubbers or other modifiers that become phase- The interaction parameter A, expressed in units of energy separated in the course of polymerization. The resulting per unit volume, is usually a positive value. This means two-phase structure exhibits an increase in the fracture that mixing occurs due to the prevailing influence of the resistance at the expense of lower values of the elastic combinatorial part of the equation over the interaction modulus and yield stress. term. Dissimilar molecules (positive A) are thus obliged Monitoring the phase separation process is important to mix due to an entropic driving force; their intrinsic in two respects: (i) for unreactive epoxy-rubber repulsion is manifested by an endothermic effect in the formulations, i.e. -

Fundamentals of Biological Mass Spectrometry and Proteomics
Fundamentals of Biological Mass Spectrometry and Proteomics Steve Carr Broad Institute of MIT and Harvard Modern Mass Spectrometer (MS) Systems Orbitrap Q-Exactive Triple Quadrupole Discovery/Global Experiments Targeted MS MS systems used for proteomics have 4 tasks: • Create ions from analyte molecules • Separate the ions based on charge and mass • Detect ions and determine their mass-to-charge • Select and fragment ions of interest to provide structural information (MS/MS) Electrospray MS: ease of coupling to liquid-based separation methods has made it the key technology in proteomics Possible Sample Inlets Syringe Pump Sample Injection Loop Liquid Autosampler, HPLC Capillary Electrophoresis Expansion of the Ion Formation and Sampling Regions Nitrogen Drying Gas Electrospray Atmosphere Vacuum Needle 3- 5 kV Liquid Nebulizing Gas Droplets Ions Containing Solvated Ions Isotopes Most elements have more than one stable isotope. For example, most carbon atoms have a mass of 12 Da, but in nature, 1.1% of C atoms have an extra neutron, making their mass 13 Da. Why do we care? Mass spectrometers “see” the isotope peaks provided the resolution is high enough. If an MS instrument has resolution high enough to resolve these isotopes, better mass accuracy is achieved. Stable isotopes of most abundant elements of peptides Element Mass Abundance H 1.0078 99.985% 2.0141 0.015 C 12.0000 98.89 13.0034 1.11 N 14.0031 99.64 15.0001 0.36 O 15.9949 99.76 16.9991 0.04 17.9992 0.20 Monoisotopic mass and isotopes We use instruments that resolve the isotopes enabling us to accurately measure the monoisotopic mass MonoisotopicMonoisotopic mass; all 12C, mass no 13C atoms corresponds to 13 lowestOne massC atom peak Two 13C atoms Angiotensin I (MW = 1295.6) (M+H)+ = C62 H90 N17 O14 TheWhen monoisotopic the isotopes mass of aare molecule clearly is the resolved sum of the the accurate monoisotopic masses for the massmost abundant isotope of each element present. -

Four Channel Liquid Chromatography/Electrochemistry
Four Channel Liquid Chromatography/Electrochemistry Bruce Peary Solomon, Ph.D. The new epsilon family of electrochemical detectors from BAS can Hong Long, Ph.D. Yongxin Zhu, Ph.D. control up to four working electrodes simultaneously. There are several Chandrani Gunaratna, Ph.D. advantages to using multiple detector electrodes. By using four different Lou Coury, Ph.D.* applied potentials with electrodes placed in a parallel arrangement, a Bioanalytical Systems, Inc. hydrodynamic voltammogram can be generated quickly through Corporate R&D Laboratories 2701 Kent Avenue acquisition of four data points for every analyte injection. This speeds West Lafayette, IN method development time. In addition, co-eluting compounds in complex 47906-1382 mixtures can be resolved on the basis of their observed half-wave * corresponding author potentials by using the same arrangement of electrodes, also in parallel. This article presents a few examples of four-electrode experiments performed with epsilon detectors in the BAS R&D labs during the past few months, using both radial-flow and cross-flow thin-layer configurations. The epsilon Platform the past fifteen years, our contract These instruments are fully network- research division, BAS Analytics, able and will be upgradable over the BAS developed and introduced the has provided analytical data of the Internet. New techniques and fea- first commercial electrochemical de- highest quality to the world’s leading tures may initially be ordered àla tector for liquid chromatography pharmaceutical companies using carte, or added at any time when the over twenty-five years ago. With this state-of-the-art products from BAS, need arises. In the coming months, issue of Current Separations,BAS as well as other leading vendors. -
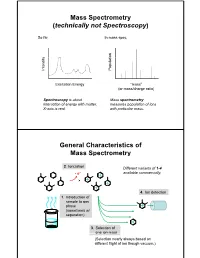
Mass Spectrometry (Technically Not Spectroscopy)
Mass Spectrometry (technically not Spectroscopy) So far, In mass spec, on y Populati Intensit Excitation Energy “mass” (or mass/charge ratio) Spectroscopy is about Mass spectrometry interaction of energy with matter. measures population of ions X-axis is real. with particular mass. General Characteristics of Mass Spectrometry 2. Ionization Different variants of 1-4 -e- available commercially. 4. Ion detection 1. Introduction of sample to gas phase (sometimes w/ separation) 3. Selection of one ion mass (Selection nearly always based on different flight of ion though vacuum.) General Components of a Mass Spectrometer Lots of choices, which can be mixed and matched. direct injection The Mass Spectrum fragment “daughter” ions M+ “parent” mass Sample Introduction: Direc t Inser tion Prob e If sample is a liquid, sample can also be injected directly into ionization region. If sample isn’t pure, get multiple parents (that can’t be distinguished from fragments). Capillary Column Introduction Continous source of molecules to spectrometer. detector column (including GC, LC, chiral, size exclusion) • Signal intensity depends on both amount of molecule and ionization efficiency • To use quantitatively, must calibrate peaks with respect eltilution time ttlitotal ion curren t to quantity eluted (TIC) over time Capillary Column Introduction Easy to interface with gas or liquid chromatography. TIC trace elution time time averaged time averaged mass spectrum mass spectrum Methods of Ionization: Electron Ionization (EI) 1 - + - 1 M + e (kV energy) M + 2e Fragmentation in Electron Ionization daughter ion (observed in spectrum) neutral fragment (not observed) excited parent at electron at electron energy of energy of 15 eV 70 e V Lower electron energy yields less fragmentation, but also less signal.