Electrochemical Real-Time Mass Spectrometry: a Novel Tool for Time-Resolved Characterization of the Products of Electrochemical Reactions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
How Do We Learn Electrochemistry? by Jeffrey W
redcat_ad_IF_Sp2012_1.pdf 1 4/11/2012 1:36:17 PM research • news • events • resources | search • explore • connect • share • discover How Do We Learn Electrochemistry? by Jeffrey W. Fergus he importance of electrochemistry is undeniable—we on education. The fall 2006 issue was devoted to education literally cannot live without electrochemistry for proper and included articles discussing general needs for education in Tcell function and transmission of signals through the electrochemistry as well as some examples of approaches, and nervous system. Electrochemistry is also vital in a wide range even specific laboratory activities, to enhance electrochemical of important technological applications. For example, batteries education. More recently, in the summer 2010 issue, the ECS are important not only in storing energy for mobile devices Industrial Electrochemistry and Electrochemical Engineering and vehicles, but also for load leveling to enable the use of Division provided an additional evaluation of needs and some renewable energy conversion technologies. Electrochemistry activities for introducing electrochemistry into courses. The is involved in the production of materials by electrorefining current issue complements those earlier issues and provides or electrodeposition as well as the destruction of materials by insights on the status and needs in electrochemical education. TM corrosion. In spite of its ubiquity there are very few formal The first article provides a general backdrop on the state educational degree programs in electrochemistry. of higher education. Marye Ann Fox discusses the financial If electrochemistry is ubiquitous, but formal educational challenges being faced by academic institutions and how these programs are rare, how do the scientists and engineers working on challenges have an impact on the design and implementation of electrochemical products and processes learn the electrochemistry educational programs. -

Gas Chromatography-Mass Spectroscopy
Gas Chromatography-Mass Spectroscopy Introduction Gas chromatography-mass spectroscopy (GC-MS) is one of the so-called hyphenated analytical techniques. As the name implies, it is actually two techniques that are combined to form a single method of analyzing mixtures of chemicals. Gas chromatography separates the components of a mixture and mass spectroscopy characterizes each of the components individually. By combining the two techniques, an analytical chemist can both qualitatively and quantitatively evaluate a solution containing a number of chemicals. Gas Chromatography In general, chromatography is used to separate mixtures of chemicals into individual components. Once isolated, the components can be evaluated individually. In all chromatography, separation occurs when the sample mixture is introduced (injected) into a mobile phase. In liquid chromatography (LC), the mobile phase is a solvent. In gas chromatography (GC), the mobile phase is an inert gas such as helium. The mobile phase carries the sample mixture through what is referred to as a stationary phase. The stationary phase is usually a chemical that can selectively attract components in a sample mixture. The stationary phase is usually contained in a tube of some sort called a column. Columns can be glass or stainless steel of various dimensions. The mixture of compounds in the mobile phase interacts with the stationary phase. Each compound in the mixture interacts at a different rate. Those that interact the fastest will exit (elute from) the column first. Those that interact slowest will exit the column last. By changing characteristics of the mobile phase and the stationary phase, different mixtures of chemicals can be separated. -

Gas-Phase Electrochemistry: Measuring Absolute Potentials and Investigating Ion and Electron Hydration*
Pure Appl. Chem., Vol. 83, No. 12, pp. 2129–2151, 2011. doi:10.1351/PAC-CON-11-08-15 © 2011 IUPAC, Publication date (Web): 7 October 2011 Gas-phase electrochemistry: Measuring absolute potentials and investigating ion and electron hydration* William A. Donald1,‡ and Evan R. Williams2 1School of Chemistry, Bio21 Institute of Molecular Science and Biotechnology, and ARC Centre of Excellence for Free Radical Chemistry and Biotechnology, University of Melbourne, Melbourne, Victoria, Australia; 2Department of Chemistry, University of California, Berkeley, CA, USA Abstract: In solution, half-cell potentials and ion solvation energies (or enthalpies) are meas- ured relative to other values, thus establishing ladders of thermochemical values that are ref- erenced to the potential of the standard hydrogen electrode (SHE) and the proton hydration energy (or enthalpy), respectively, which are both arbitrarily assigned a value of 0. In this focused review article, we describe three routes for obtaining absolute solution-phase half- cell potentials using ion nanocalorimetry, in which the energy resulting from electron capture (EC) by large hydrated ions in the gas phase are obtained from the number of water mole- cules lost from the reduced precursor cluster, which was developed by the Williams group at the University of California, Berkeley. Recent ion nanocalorimetry methods for investigating ion and electron hydration and for obtaining the absolute hydration enthalpy of the electron are discussed. From these methods, an absolute electrochemical scale and ion solvation scale can be established from experimental measurements without any models. Keywords: absolute potentials; clusters; electrochemistry; electron capture dissociation; elec- tron transfer; gaseous state; hydrated ions; hydration; ion nanocalorimetry; solvation; thermo chemistry. -

Ultra-Performance Liquid Chromatography Coupled to Quadrupole-Orthogonal Time-Of-flight Mass Spectrometry
RAPID COMMUNICATIONS IN MASS SPECTROMETRY Rapid Commun. Mass Spectrom. 2004; 18: 2331–2337 Published online in Wiley InterScience (www.interscience.wiley.com). DOI: 10.1002/rcm.1627 Ultra-performance liquid chromatography coupled to quadrupole-orthogonal time-of-flight mass spectrometry Robert Plumb1*, Jose Castro-Perez2, Jennifer Granger1, Iain Beattie3, Karine Joncour3 and Andrew Wright3 1Waters Corporation, Milford, MA, USA 2Waters Corporation, MS Technology Center, Manchester, UK 3AstraZeneca R&D Charnwood, Physical & Metabolic Science, Loughborough, UK Received 19 June 2004; Revised 5 August 2004; Accepted 9 August 2004 Ultra-performance liquid chromatography (UPLC) utilizes sub-2 mm particles with high linear sol- vent velocities to effect dramatic increases in resolution, sensitivity and speed of analysis. The reduction in particle size to below 2 mm requires instrumentation that can operate at pressures in the 6000–15 000 psi range. The typical peak widths generated by the UPLC system are in the order of 1–2 s for a 10-min separation. In the present work this technology has been applied to the study of in vivo drug metabolism, in particular the analysis of drug metabolites in bile. The reduction in peak width significantly increases analytical sensitivity by three- to five-fold, and the reduction in peak width, and concomitant increase in peak capacity, significantly reduces spectral overlap resulting in superior spectral quality in both MS and MS/MS modes. The application of UPLC/ MS resulted in the detection of additional drug metabolites, superior separation and improved spectral quality. Copyright # 2004 John Wiley & Sons, Ltd. The detection and identification of drug metabolites is crucial Liquid chromatography/mass spectrometry (LC/MS) and to both the drug discovery and development processes, LC/MS/MS have become the mainstay of the drug metabo- although in these two areas the emphasis is slightly different. -

Electrochemistry Cell Model
Electrochemistry Cell Model Dennis Dees and Kevin Gallagher Chemical Sciences and Engineering Division May 9-13, 2011 Vehicle Technologies Program Annual Merit Review and Peer Evaluation Meeting Washington, D.C. Project ID: ES031 This presentation does not contain any proprietary, confidential, or otherwise restricted information. Overview Timeline Barriers . Start: October 2008 . Development of a safe cost-effective PHEV . Finish: September 2014 battery with a 40 mile all electric range . ~43% Complete that meets or exceeds all performance goals – Interpreting complex cell electrochemical phenomena – Identification of cell degradation mechanisms Budget Partners (Collaborators) . Total project funding . Daniel Abraham, Argonne – 100% DOE . Sun-Ho Kang, Argonne . FY2010: $400K . Andrew Jansen, Argonne . FY2011: $400K . Wenquan Lu, Argonne . Kevin Gering, INL Vehicle Technologies Program 2 Objectives, Milestones, and Approach . The objective of this work is to correlate analytical diagnostic results with the electrochemical performance of advanced lithium-ion battery technologies for PHEV applications – Link experimental efforts through electrochemical modeling studies – Identify performance limitations and aging mechanisms . Milestones for this year: – Initiate development of AC impedance two phase model (completed) – Integrate SEI growth model into full cell model (completed) . Approach for electrochemical modeling activities is to build on earlier successful characterization and modeling studies in extending efforts to new PHEV technologies -

Coupling Gas Chromatography to Mass Spectrometry
Coupling Gas Chromatography to Mass Spectrometry Introduction The suite of gas chromatographic detectors includes (roughly in order from most common to the least): the flame ionization detector (FID), thermal conductivity detector (TCD or hot wire detector), electron capture detector (ECD), photoionization detector (PID), flame photometric detector (FPD), thermionic detector, and a few more unusual or VERY expensive choices like the atomic emission detector (AED) and the ozone- or fluorine-induce chemiluminescence detectors. All of these except the AED produce an electrical signal that varies with the amount of analyte exiting the chromatographic column. The AED does that AND yields the emission spectrum of selected elements in the analytes as well. Another GC detector that is also very expensive but very powerful is a scaled down version of the mass spectrometer. When coupled to a GC the detection system itself is often referred to as the mass selective detector or more simply the mass detector. This powerful analytical technique belongs to the class of hyphenated analytical instrumentation (since each part had a different beginning and can exist independently) and is called gas chromatograhy/mass spectrometry (GC/MS). Placed at the end of a capillary column in a manner similar to the other GC detectors, the mass detector is more complicated than, for instance, the FID because of the mass spectrometer's complex requirements for the process of creation, separation, and detection of gas phase ions. A capillary column is required in the chromatograph because the entire MS process must be carried out at very low pressures (~10-5 torr) and in order to meet this requirement a vacuum is maintained via constant pumping using a vacuum pump. -

Physical and Analytical Electrochemistry: the Fundamental
Electrochemical Systems The simplest and traditional electrochemical process occurring at the boundary between an electronically conducting phase (the electrode) and an ionically conducting phase (the electrolyte solution), is the heterogeneous electro-transfer step between the electrode and the electroactive species of interest present in the solution. An example is the plating of nickel. Ni2+ + 2e- → Ni Physical and Analytical The interface is where the action occurs but connected to that central event are various processes that can Electrochemistry: occur in parallel or series. Figure 2 demonstrates a more complex interface; it represents a molecular scale snapshot The Fundamental Core of an interface that exists in a fuel cell with a solid polymer (capable of conducting H+) as an electrolyte. of Electrochemistry However, there is more to an electrochemical system than a single by Tom Zawodzinski, Shelley Minteer, interface. An entire circuit must be made and Gessie Brisard for measurable current to flow. This circuit consists of the electrochemical cell plus external wiring and circuitry The common event for all electrochemical processes is that (power sources, measuring devices, of electron transfer between chemical species; or between an etc). The cell consists of (at least) electrode and a chemical species situated in the vicinity of the two electrodes separated by (at least) one electrolyte solution. Figure 3 is electrode, usually a pure metal or an alloy. The location where a schematic of a simple circuit. Each the electron transfer reactions take place is of fundamental electrode has an interface with a importance in electrochemistry because it regulates the solution. Electrons flow in the external behavior of most electrochemical systems. -

Concepts and Tools for Mechanism and Selectivity Analysis in Synthetic Organic Electrochemistry
Concepts and tools for mechanism and selectivity analysis in synthetic organic electrochemistry Cyrille Costentina,1,2 and Jean-Michel Savéanta,1 aUniversité Paris Diderot, Sorbonne Paris Cité, Laboratoire d’Electrochimie Moléculaire, Unité Mixte de Recherche Université–CNRS 7591, 75205 Paris Cedex 13, France Contributed by Jean-Michel Savéant, April 2, 2019 (sent for review March 19, 2019; reviewed by Robert Francke and R. Daniel Little) As an accompaniment to the current renaissance of synthetic organic sufficient to record a current-potential response but small electrochemistry, the heterogeneous and space-dependent nature of enough to leave the substrates and cosubstrates (of the order of electrochemical reactions is analyzed in detail. The reactions that follow one part per million) almost untouched. Competition of the the initial electron transfer step and yield the products are intimately electrochemical/chemical events with diffusional transport under coupled with reactant transport. Depiction of the ensuing reactions precisely mastered conditions allows analysis of the kinetics profiles is the key to the mechanism and selectivity parameters. within extended time windows (from minutes to submicroseconds). Analysis is eased by the steady state resulting from coupling of However, for irreversible processes, these approaches are blind on diffusion with convection forced by solution stirring or circulation. reaction bifurcations occurring beyond the kinetically determining Homogeneous molecular catalysis of organic electrochemical reactions step, which are precisely those governing the selectivity of the re- of the redox or chemical type may be treated in the same manner. The same benchmarking procedures recently developed for the activation action. This is not the case of preparative-scale electrolysis accom- of small molecules in the context of modern energy challenges lead to panied by identification and quantitation of products. -

Electron Ionization
Chapter 6 Chapter 6 Electron Ionization I. Introduction ......................................................................................................317 II. Ionization Process............................................................................................317 III. Strategy for Data Interpretation......................................................................321 1. Assumptions 2. The Ionization Process IV. Types of Fragmentation Pathways.................................................................328 1. Sigma-Bond Cleavage 2. Homolytic or Radical-Site-Driven Cleavage 3. Heterolytic or Charge-Site-Driven Cleavage 4. Rearrangements A. Hydrogen-Shift Rearrangements B. Hydride-Shift Rearrangements V. Representative Fragmentations (Spectra) of Classes of Compounds.......... 344 1. Hydrocarbons A. Saturated Hydrocarbons 1) Straight-Chain Hydrocarbons 2) Branched Hydrocarbons 3) Cyclic Hydrocarbons B. Unsaturated C. Aromatic 2. Alkyl Halides 3. Oxygen-Containing Compounds A. Aliphatic Alcohols B. Aliphatic Ethers C. Aromatic Alcohols D. Cyclic Ethers E. Ketones and Aldehydes F. Aliphatic Acids and Esters G. Aromatic Acids and Esters 4. Nitrogen-Containing Compounds A. Aliphatic Amines B. Aromatic Compounds Containing Atoms of Nitrogen C. Heterocyclic Nitrogen-Containing Compounds D. Nitro Compounds E. Concluding Remarks on the Mass Spectra of Nitrogen-Containing Compounds 5. Multiple Heteroatoms or Heteroatoms and a Double Bond 6. Trimethylsilyl Derivative 7. Determining the Location of Double Bonds VI. Library -

Fundamentals of Biological Mass Spectrometry and Proteomics
Fundamentals of Biological Mass Spectrometry and Proteomics Steve Carr Broad Institute of MIT and Harvard Modern Mass Spectrometer (MS) Systems Orbitrap Q-Exactive Triple Quadrupole Discovery/Global Experiments Targeted MS MS systems used for proteomics have 4 tasks: • Create ions from analyte molecules • Separate the ions based on charge and mass • Detect ions and determine their mass-to-charge • Select and fragment ions of interest to provide structural information (MS/MS) Electrospray MS: ease of coupling to liquid-based separation methods has made it the key technology in proteomics Possible Sample Inlets Syringe Pump Sample Injection Loop Liquid Autosampler, HPLC Capillary Electrophoresis Expansion of the Ion Formation and Sampling Regions Nitrogen Drying Gas Electrospray Atmosphere Vacuum Needle 3- 5 kV Liquid Nebulizing Gas Droplets Ions Containing Solvated Ions Isotopes Most elements have more than one stable isotope. For example, most carbon atoms have a mass of 12 Da, but in nature, 1.1% of C atoms have an extra neutron, making their mass 13 Da. Why do we care? Mass spectrometers “see” the isotope peaks provided the resolution is high enough. If an MS instrument has resolution high enough to resolve these isotopes, better mass accuracy is achieved. Stable isotopes of most abundant elements of peptides Element Mass Abundance H 1.0078 99.985% 2.0141 0.015 C 12.0000 98.89 13.0034 1.11 N 14.0031 99.64 15.0001 0.36 O 15.9949 99.76 16.9991 0.04 17.9992 0.20 Monoisotopic mass and isotopes We use instruments that resolve the isotopes enabling us to accurately measure the monoisotopic mass MonoisotopicMonoisotopic mass; all 12C, mass no 13C atoms corresponds to 13 lowestOne massC atom peak Two 13C atoms Angiotensin I (MW = 1295.6) (M+H)+ = C62 H90 N17 O14 TheWhen monoisotopic the isotopes mass of aare molecule clearly is the resolved sum of the the accurate monoisotopic masses for the massmost abundant isotope of each element present. -

Four Channel Liquid Chromatography/Electrochemistry
Four Channel Liquid Chromatography/Electrochemistry Bruce Peary Solomon, Ph.D. The new epsilon family of electrochemical detectors from BAS can Hong Long, Ph.D. Yongxin Zhu, Ph.D. control up to four working electrodes simultaneously. There are several Chandrani Gunaratna, Ph.D. advantages to using multiple detector electrodes. By using four different Lou Coury, Ph.D.* applied potentials with electrodes placed in a parallel arrangement, a Bioanalytical Systems, Inc. hydrodynamic voltammogram can be generated quickly through Corporate R&D Laboratories 2701 Kent Avenue acquisition of four data points for every analyte injection. This speeds West Lafayette, IN method development time. In addition, co-eluting compounds in complex 47906-1382 mixtures can be resolved on the basis of their observed half-wave * corresponding author potentials by using the same arrangement of electrodes, also in parallel. This article presents a few examples of four-electrode experiments performed with epsilon detectors in the BAS R&D labs during the past few months, using both radial-flow and cross-flow thin-layer configurations. The epsilon Platform the past fifteen years, our contract These instruments are fully network- research division, BAS Analytics, able and will be upgradable over the BAS developed and introduced the has provided analytical data of the Internet. New techniques and fea- first commercial electrochemical de- highest quality to the world’s leading tures may initially be ordered àla tector for liquid chromatography pharmaceutical companies using carte, or added at any time when the over twenty-five years ago. With this state-of-the-art products from BAS, need arises. In the coming months, issue of Current Separations,BAS as well as other leading vendors. -
Mass Spectrometry (Technically Not Spectroscopy)
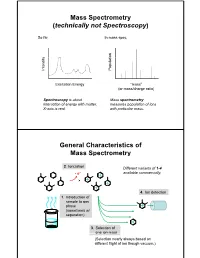
Mass Spectrometry (technically not Spectroscopy) So far, In mass spec, on y Populati Intensit Excitation Energy “mass” (or mass/charge ratio) Spectroscopy is about Mass spectrometry interaction of energy with matter. measures population of ions X-axis is real. with particular mass. General Characteristics of Mass Spectrometry 2. Ionization Different variants of 1-4 -e- available commercially. 4. Ion detection 1. Introduction of sample to gas phase (sometimes w/ separation) 3. Selection of one ion mass (Selection nearly always based on different flight of ion though vacuum.) General Components of a Mass Spectrometer Lots of choices, which can be mixed and matched. direct injection The Mass Spectrum fragment “daughter” ions M+ “parent” mass Sample Introduction: Direc t Inser tion Prob e If sample is a liquid, sample can also be injected directly into ionization region. If sample isn’t pure, get multiple parents (that can’t be distinguished from fragments). Capillary Column Introduction Continous source of molecules to spectrometer. detector column (including GC, LC, chiral, size exclusion) • Signal intensity depends on both amount of molecule and ionization efficiency • To use quantitatively, must calibrate peaks with respect eltilution time ttlitotal ion curren t to quantity eluted (TIC) over time Capillary Column Introduction Easy to interface with gas or liquid chromatography. TIC trace elution time time averaged time averaged mass spectrum mass spectrum Methods of Ionization: Electron Ionization (EI) 1 - + - 1 M + e (kV energy) M + 2e Fragmentation in Electron Ionization daughter ion (observed in spectrum) neutral fragment (not observed) excited parent at electron at electron energy of energy of 15 eV 70 e V Lower electron energy yields less fragmentation, but also less signal.