Ch.14-16 Electrochemistry Redox Reaction
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chemistry Grade Level 10 Units 1-15
COPPELL ISD SUBJECT YEAR AT A GLANCE GRADE HEMISTRY UNITS C LEVEL 1-15 10 Program Transfer Goals ● Ask questions, recognize and define problems, and propose solutions. ● Safely and ethically collect, analyze, and evaluate appropriate data. ● Utilize, create, and analyze models to understand the world. ● Make valid claims and informed decisions based on scientific evidence. ● Effectively communicate scientific reasoning to a target audience. PACING 1st 9 Weeks 2nd 9 Weeks 3rd 9 Weeks 4th 9 Weeks Unit 1 Unit 2 Unit 3 Unit 4 Unit 5 Unit 6 Unit Unit Unit Unit Unit Unit Unit Unit Unit 7 8 9 10 11 12 13 14 15 1.5 wks 2 wks 1.5 wks 2 wks 3 wks 5.5 wks 1.5 2 2.5 2 wks 2 2 2 wks 1.5 1.5 wks wks wks wks wks wks wks Assurances for a Guaranteed and Viable Curriculum Adherence to this scope and sequence affords every member of the learning community clarity on the knowledge and skills on which each learner should demonstrate proficiency. In order to deliver a guaranteed and viable curriculum, our team commits to and ensures the following understandings: Shared Accountability: Responding -

Chapter 9 Titrimetric Methods 413
Chapter 9 Titrimetric Methods Chapter Overview Section 9A Overview of Titrimetry Section 9B Acid–Base Titrations Section 9C Complexation Titrations Section 9D Redox Titrations Section 9E Precipitation Titrations Section 9F Key Terms Section 9G Chapter Summary Section 9H Problems Section 9I Solutions to Practice Exercises Titrimetry, in which volume serves as the analytical signal, made its first appearance as an analytical method in the early eighteenth century. Titrimetric methods were not well received by the analytical chemists of that era because they could not duplicate the accuracy and precision of a gravimetric analysis. Not surprisingly, few standard texts from the 1700s and 1800s include titrimetric methods of analysis. Precipitation gravimetry developed as an analytical method without a general theory of precipitation. An empirical relationship between a precipitate’s mass and the mass of analyte— what analytical chemists call a gravimetric factor—was determined experimentally by taking a known mass of analyte through the procedure. Today, we recognize this as an early example of an external standardization. Gravimetric factors were not calculated using the stoichiometry of a precipitation reaction because chemical formulas and atomic weights were not yet available! Unlike gravimetry, the development and acceptance of titrimetry required a deeper understanding of stoichiometry, of thermodynamics, and of chemical equilibria. By the 1900s, the accuracy and precision of titrimetric methods were comparable to that of gravimetric methods, establishing titrimetry as an accepted analytical technique. 411 412 Analytical Chemistry 2.0 9A Overview of Titrimetry We are deliberately avoiding the term In titrimetry we add a reagent, called the titrant, to a solution contain- analyte at this point in our introduction ing another reagent, called the titrand, and allow them to react. -

Nernst Equation in Electrochemistry the Nernst Equation Gives the Reduction Potential of a Half‐Cell in Equilibrium
Nernst equation In electrochemistry the Nernst equation gives the reduction potential of a half‐cell in equilibrium. In further cases, it can also be used to determine the emf (electromotive force) for a full electrochemical cell. (half‐cell reduction potential) (total cell potential) where Ered is the half‐cell reduction potential at a certain T o E red is the standard half‐cell reduction potential Ecell is the cell potential (electromotive force) o E cell is the standard cell potential at a certain T R is the universal gas constant: R = 8.314472(15) JK−1mol−1 T is the absolute temperature in Kelvin a is the chemical activity for the relevant species, where aRed is the reductant and aOx is the oxidant F is the Faraday constant; F = 9.64853399(24)×104 Cmol−1 z is the number of electrons transferred in the cell reaction or half‐reaction Q is the reaction quotient (e.g. molar concentrations, partial pressures …) As the system is considered not to have reached equilibrium, the reaction quotient Q is used instead of the equilibrium constant k. The electrochemical series is used to determine the electrochemical potential or the electrode potential of an electrochemical cell. These electrode potentials are measured relatively to the standard hydrogen electrode. A reduced member of a couple has a thermodynamic tendency to reduce the oxidized member of any couple that lies above it in the series. The standard hydrogen electrode is a redox electrode which forms the basis of the thermodynamic scale of these oxidation‐ reduction potentials. For a comparison with all other electrode reactions, standard electrode potential E0 of hydrogen is defined to be zero at all temperatures. -

Titration Endpoint Challenge
Analytical and Bioanalytical Chemistry (2019) 411:1–2 https://doi.org/10.1007/s00216-018-1430-y ANALYTICAL CHALLENGE Titration endpoint challenge Diego Alejandro Ahumada Forigua1 & Juris Meija2 Published online: 1 January 2019 # Springer-Verlag GmbH Germany, part of Springer Nature 2018 We would like to invite you to participate in the Analytical practical aspects related to the sources of uncertainty of Challenge, a series of puzzles to entertain and challenge our this technique. readers. This special feature of “Analytical and Bioanalytical Origins of titrimetry date back to 1690s, when Wilhelm Chemistry” has established itself as a truly unique quiz series, Homberg (1652–1715) published the first report related to with a new scientific puzzle published every three months. an acidity measurement [1]. Several decades later Claude Readers can access the complete collection of published prob- Geoffroy (1729–1753) used this method to determine the lems with their solutions on the ABC homepage at http://www. strength of vinegar by adding small amounts of potassium springer.com/abc. Test your knowledge and tease your wits in carbonate until the no further effervescence was observed diverse areas of analytical and bioanalytical chemistry by [2]. William Lewis (1708–1781), who is also considered one viewing this collection. of the early pioneers of titration, recognized the difficulty in In the present challenge, titration is the topic. And please determining the endpoint of the titration through the process note that there is a prize to be won (a Springer book of your of cessation of effervescence, so he suggested the use of color choice up to a value of €100). -

Chapter 3: Experimental
Chapter 3: Experimental CHAPTER 3: EXPERIMENTAL 3.1 Basic concepts of the experimental techniques In this part of the chapter, a short overview of some phrases and theoretical aspects of the experimental techniques used in this work are given. Cyclic voltammetry (CV) is the most common technique to obtain preliminary information about an electrochemical process. It is sensitive to the mechanism of deposition and therefore provides informations on structural transitions, as well as interactions between the surface and the adlayer. Chronoamperometry is very powerful method for the quantitative analysis of a nucleation process. The scanning tunneling microscopy (STM) is based on the exponential dependence of the tunneling current, flowing from one electrode onto another one, depending on the distance between electrodes. Combination of the STM with an electrochemical cell allows in-situ study of metal electrochemical phase formation. XPS is also a very powerfull technique to investigate the chemical states of adsorbates. Theoretical background of these techniques will be given in the following pages. At an electrode surface, two fundamental electrochemical processes can be distinguished: 3.1.1 Capacitive process Capacitive processes are caused by the (dis-)charge of the electrode surface as a result of a potential variation, or by an adsorption process. Capacitive current, also called "non-faradaic" or "double-layer" current, does not involve any chemical reactions (charge transfer), it only causes accumulation (or removal) of electrical charges on the electrode and in the electrolyte solution near the electrode. There is always some capacitive current flowing when the potential of an electrode is changing. In contrast to faradaic current, capacitive current can also flow at constant 28 Chapter 3: Experimental potential if the capacitance of the electrode is changing for some reason, e.g., change of electrode area, adsorption or temperature. -
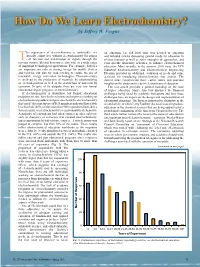
How Do We Learn Electrochemistry? by Jeffrey W
redcat_ad_IF_Sp2012_1.pdf 1 4/11/2012 1:36:17 PM research • news • events • resources | search • explore • connect • share • discover How Do We Learn Electrochemistry? by Jeffrey W. Fergus he importance of electrochemistry is undeniable—we on education. The fall 2006 issue was devoted to education literally cannot live without electrochemistry for proper and included articles discussing general needs for education in Tcell function and transmission of signals through the electrochemistry as well as some examples of approaches, and nervous system. Electrochemistry is also vital in a wide range even specific laboratory activities, to enhance electrochemical of important technological applications. For example, batteries education. More recently, in the summer 2010 issue, the ECS are important not only in storing energy for mobile devices Industrial Electrochemistry and Electrochemical Engineering and vehicles, but also for load leveling to enable the use of Division provided an additional evaluation of needs and some renewable energy conversion technologies. Electrochemistry activities for introducing electrochemistry into courses. The is involved in the production of materials by electrorefining current issue complements those earlier issues and provides or electrodeposition as well as the destruction of materials by insights on the status and needs in electrochemical education. TM corrosion. In spite of its ubiquity there are very few formal The first article provides a general backdrop on the state educational degree programs in electrochemistry. of higher education. Marye Ann Fox discusses the financial If electrochemistry is ubiquitous, but formal educational challenges being faced by academic institutions and how these programs are rare, how do the scientists and engineers working on challenges have an impact on the design and implementation of electrochemical products and processes learn the electrochemistry educational programs. -

Square Wave Voltammetry
Application Note S-7 Subject: Square Wave Voltammetry INTRODUCTION reverse pulse of the square wave occurs half way through the staircase step. The primary advantage of the pulse voltammetric techniques, such as normal pulse or differential pulse voltammetry, is their ability to discriminate against charging (capacitance) current. As a result, the pulse techniques are more sensitive to oxidation or reduction currents (faradaic currents) than conventional dc PLUS voltammetry. Differential pulse voltammetry yields peaks for faradaic currents rather than the sigmoidal waveform obtained with dc or normal pulse techniques. This results in improved resolution for multiple analyte systems and more convenient quantitation. Square wave voltammetry has received growing attention as a voltammetric technique for routine quantitative analyses. Although square wave voltammetry was E reported as long ago as 1957 by Barker1, the utility of the EQUALS technique was limited by electronic technology. Recent advances in both analog and digital electronics have made it feasible to incorporate square wave voltammetry into the PAR Model 394 Polarographic Analyzer. Square wave voltammetry can be used to perform an experiment much faster than normal and differential pulse techniques, which typically run at scan rates of 1 to 10 mV/sec. Square wave voltammetry employs scan rates TIME up to 1 V/sec or faster, allowing much faster determinations. A typical experiment requiring three minutes by normal or differential pulse techniques can be performed in a matter of seconds by square wave voltammetry. FIGURE 1: Applied excitation in square wave voltammetry. THEORY The timing and applied potential parameters for square The waveform used for square wave voltammetry is wave voltammetry are depicted in Figure 2. -

Electrochemical Real-Time Mass Spectrometry: a Novel Tool for Time-Resolved Characterization of the Products of Electrochemical Reactions
Electrochemical real-time mass spectrometry: A novel tool for time-resolved characterization of the products of electrochemical reactions Elektrochemische Realzeit-Massenspektrometrie: Eine neuartige Methode zur zeitaufgelösten Charakterisierung der Produkte elektrochemischer Reaktionen Der Technischen Fakultät der Friedrich-Alexander-Universität Erlangen-Nürnberg zur Erlangung des Doktorgrades Dr.-Ingenieur vorgelegt von Peyman Khanipour Mehrin aus Shiraz, Iran Als Dissertation genehmigt von der Technischen Fakultät der Friedrich-Alexander-Universität Erlangen-Nürnberg Tag der mündlichen Prüfung: 17.11.2020 Vorsitzender des Promotionsorgans: Prof. Dr.-Ing. habil. Andreas Paul Fröba Gutachter: Prof. Dr. Karl J.J. Mayrhofer Prof. Dr. Frank-Michael Matysik I Acknowledgements This study is done in the electrosynthesis team of the electrocatalysis unit at Helmholtz- Institut Erlangen-Nürnberg (HI ERN) with the financial support of Forschungszentrum Jülich. I would like to express my deep gratitude to Prof. Dr. Karl J. J. Mayrhofer for accepting me as a Ph.D. student and also for all his encouragement, supports, and freedoms during my study. I’m grateful to Prof. Dr. Frank-Michael Matysik for kindly accepting to act as a second reviewer and also for the time he has invested in reading this thesis. This piece of work is enabled by collaboration with scientists from different expertise. I would like to express my appreciation to Dr. Sandra Haschke from FAU for providing shape-controlled high surface area platinum electrodes which I used for performing oxidation of primary alcohols and also the characterization of the provided material SEM, EDX, and XRD. Mr. Mario Löffler from HI ERN for obtaining the XPS data and his remarkable knowledge with the interpretation of the spectra on copper-based electrodes for the CO 2 electroreduction reaction. -

Gas-Phase Electrochemistry: Measuring Absolute Potentials and Investigating Ion and Electron Hydration*
Pure Appl. Chem., Vol. 83, No. 12, pp. 2129–2151, 2011. doi:10.1351/PAC-CON-11-08-15 © 2011 IUPAC, Publication date (Web): 7 October 2011 Gas-phase electrochemistry: Measuring absolute potentials and investigating ion and electron hydration* William A. Donald1,‡ and Evan R. Williams2 1School of Chemistry, Bio21 Institute of Molecular Science and Biotechnology, and ARC Centre of Excellence for Free Radical Chemistry and Biotechnology, University of Melbourne, Melbourne, Victoria, Australia; 2Department of Chemistry, University of California, Berkeley, CA, USA Abstract: In solution, half-cell potentials and ion solvation energies (or enthalpies) are meas- ured relative to other values, thus establishing ladders of thermochemical values that are ref- erenced to the potential of the standard hydrogen electrode (SHE) and the proton hydration energy (or enthalpy), respectively, which are both arbitrarily assigned a value of 0. In this focused review article, we describe three routes for obtaining absolute solution-phase half- cell potentials using ion nanocalorimetry, in which the energy resulting from electron capture (EC) by large hydrated ions in the gas phase are obtained from the number of water mole- cules lost from the reduced precursor cluster, which was developed by the Williams group at the University of California, Berkeley. Recent ion nanocalorimetry methods for investigating ion and electron hydration and for obtaining the absolute hydration enthalpy of the electron are discussed. From these methods, an absolute electrochemical scale and ion solvation scale can be established from experimental measurements without any models. Keywords: absolute potentials; clusters; electrochemistry; electron capture dissociation; elec- tron transfer; gaseous state; hydrated ions; hydration; ion nanocalorimetry; solvation; thermo chemistry. -

Elements of Electrochemistry
Page 1 of 8 Chem 201 Winter 2006 ELEM ENTS OF ELEC TROCHEMIS TRY I. Introduction A. A number of analytical techniques are based upon oxidation-reduction reactions. B. Examples of these techniques would include: 1. Determinations of Keq and oxidation-reduction midpoint potentials. 2. Determination of analytes by oxidation-reductions titrations. 3. Ion-specific electrodes (e.g., pH electrodes, etc.) 4. Gas-sensing probes. 5. Electrogravimetric analysis: oxidizing or reducing analytes to a known product and weighing the amount produced 6. Coulometric analysis: measuring the quantity of electrons required to reduce/oxidize an analyte II. Terminology A. Reduction: the gaining of electrons B. Oxidation: the loss of electrons C. Reducing agent (reductant): species that donates electrons to reduce another reagent. (The reducing agent get oxidized.) D. Oxidizing agent (oxidant): species that accepts electrons to oxidize another species. (The oxidizing agent gets reduced.) E. Oxidation-reduction reaction (redox reaction): a reaction in which electrons are transferred from one reactant to another. 1. For example, the reduction of cerium(IV) by iron(II): Ce4+ + Fe2+ ! Ce3+ + Fe3+ a. The reduction half-reaction is given by: Ce4+ + e- ! Ce3+ b. The oxidation half-reaction is given by: Fe2+ ! e- + Fe3+ 2. The half-reactions are the overall reaction broken down into oxidation and reduction steps. 3. Half-reactions cannot occur independently, but are used conceptually to simplify understanding and balancing the equations. III. Rules for Balancing Oxidation-Reduction Reactions A. Write out half-reaction "skeletons." Page 2 of 8 Chem 201 Winter 2006 + - B. Balance the half-reactions by adding H , OH or H2O as needed, maintaining electrical neutrality. -

Electrochemistry Cell Model
Electrochemistry Cell Model Dennis Dees and Kevin Gallagher Chemical Sciences and Engineering Division May 9-13, 2011 Vehicle Technologies Program Annual Merit Review and Peer Evaluation Meeting Washington, D.C. Project ID: ES031 This presentation does not contain any proprietary, confidential, or otherwise restricted information. Overview Timeline Barriers . Start: October 2008 . Development of a safe cost-effective PHEV . Finish: September 2014 battery with a 40 mile all electric range . ~43% Complete that meets or exceeds all performance goals – Interpreting complex cell electrochemical phenomena – Identification of cell degradation mechanisms Budget Partners (Collaborators) . Total project funding . Daniel Abraham, Argonne – 100% DOE . Sun-Ho Kang, Argonne . FY2010: $400K . Andrew Jansen, Argonne . FY2011: $400K . Wenquan Lu, Argonne . Kevin Gering, INL Vehicle Technologies Program 2 Objectives, Milestones, and Approach . The objective of this work is to correlate analytical diagnostic results with the electrochemical performance of advanced lithium-ion battery technologies for PHEV applications – Link experimental efforts through electrochemical modeling studies – Identify performance limitations and aging mechanisms . Milestones for this year: – Initiate development of AC impedance two phase model (completed) – Integrate SEI growth model into full cell model (completed) . Approach for electrochemical modeling activities is to build on earlier successful characterization and modeling studies in extending efforts to new PHEV technologies -

Reaction Engineering of Polymer Electrolyte Membrane Fuel Cells
Reaction Engineering of Polymer Electrolyte Membrane Fuel Cells A new approach to elucidate the operation and control of Polymer Electrolyte Membrane (PEM) fuel cells is being developed. A global reactor engineering approach is applied to PEM fuel cells to identify the essential physics that govern the dynamics in PEM fuel cells. Reaction engineering principles are employed to develop a one-dimensional differential PEM fuel cell suitable for elucidating the dynamic performance of PEM cells under well-defined conditions. Polymer Electrolyte Fuel Cells Polymer electrolyte membrane (PEM) fuel cells employ a polymer membrane with acid side groups to conduct protons from the anode to cathode. Water management in the fuel cell is critical for PEM fuel cell operation. Sufficient water must be absorbed into the membrane to ionize the acid groups; excess water can flood the cathode of the fuel cell diminishing fuel cell performance limiting the power output. A schematic of a polymer electrolyte membrane hydrogen-oxygen fuel cell is shown in Figure 1. load e- Figure 1. Hydrogen-oxygen PEM fuel cell. Hydrogen molecules hydrogen in oxygen in dissociatively adsorb at the anode and are oxidized to protons. Electrons travel through an external load resistance. Protons diffuse H+ through the PEM under an e t electrochemical gradient to the y l ro t c cathode. Oxygen molecules adsorb e l at the cathode, are reduced and react hydrogen r E oxygen e m y + water out with the protons to produce water. + water out l Po The product water is absorbed into the PEM, or evaporates into the gas anode cathode streams at the anode and cathode.