Fundamentals of Biological Mass Spectrometry and Proteomics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

An Introduction to Isotopic Calculations John M
An Introduction to Isotopic Calculations John M. Hayes ([email protected]) Woods Hole Oceanographic Institution, Woods Hole, MA 02543, USA, 30 September 2004 Abstract. These notes provide an introduction to: termed isotope effects. As a result of such effects, the • Methods for the expression of isotopic abundances, natural abundances of the stable isotopes of practically • Isotopic mass balances, and all elements involved in low-temperature geochemical • Isotope effects and their consequences in open and (< 200°C) and biological processes are not precisely con- closed systems. stant. Taking carbon as an example, the range of interest is roughly 0.00998 ≤ 13F ≤ 0.01121. Within that range, Notation. Absolute abundances of isotopes are com- differences as small as 0.00001 can provide information monly reported in terms of atom percent. For example, about the source of the carbon and about processes in 13 13 12 13 atom percent C = [ C/( C + C)]100 (1) which the carbon has participated. A closely related term is the fractional abundance The delta notation. Because the interesting isotopic 13 13 fractional abundance of C ≡ F differences between natural samples usually occur at and 13F = 13C/(12C + 13C) (2) beyond the third significant figure of the isotope ratio, it has become conventional to express isotopic abundances These variables deserve attention because they provide using a differential notation. To provide a concrete the only basis for perfectly accurate mass balances. example, it is far easier to say – and to remember – that Isotope ratios are also measures of the absolute abun- the isotope ratios of samples A and B differ by one part dance of isotopes; they are usually arranged so that the per thousand than to say that sample A has 0.3663 %15N more abundant isotope appears in the denominator and sample B has 0.3659 %15N. -

Mass Spectrometry: Quadrupole Mass Filter
Advanced Lab, Jan. 2008 Mass Spectrometry: Quadrupole Mass Filter The mass spectrometer is essentially an instrument which can be used to measure the mass, or more correctly the mass/charge ratio, of ionized atoms or other electrically charged particles. Mass spectrometers are now used in physics, geology, chemistry, biology and medicine to determine compositions, to measure isotopic ratios, for detecting leaks in vacuum systems, and in homeland security. Mass Spectrometer Designs The first mass spectrographs were invented almost 100 years ago, by A.J. Dempster, F.W. Aston and others, and have therefore been in continuous development over a very long period. However the principle of using electric and magnetic fields to accelerate and establish the trajectories of ions inside the spectrometer according to their mass/charge ratio is common to all the different designs. The following description of Dempster’s original mass spectrograph is a simple illustration of these physical principles: The magnetic sector spectrograph PUMP F DD S S3 1 r S2 Fig. 1: Dempster’s Mass Spectrograph (1918). Atoms/molecules are first ionized by electrons emitted from the hot filament (F) and then accelerated towards the entrance slit (S1). The ions then follow a semicircular trajectory established by the Lorentz force in a uniform magnetic field. The radius of the trajectory, r, is defined by three slits (S1, S2, and S3). Ions with this selected trajectory are then detected by the detector D. How the magnetic sector mass spectrograph works: Equating the Lorentz force with the centripetal force gives: qvB = mv2/r (1) where q is the charge on the ion (usually +e), B the magnetic field, m is the mass of the ion and r the radius of the ion trajectory. -

Gas Chromatography-Mass Spectroscopy
Gas Chromatography-Mass Spectroscopy Introduction Gas chromatography-mass spectroscopy (GC-MS) is one of the so-called hyphenated analytical techniques. As the name implies, it is actually two techniques that are combined to form a single method of analyzing mixtures of chemicals. Gas chromatography separates the components of a mixture and mass spectroscopy characterizes each of the components individually. By combining the two techniques, an analytical chemist can both qualitatively and quantitatively evaluate a solution containing a number of chemicals. Gas Chromatography In general, chromatography is used to separate mixtures of chemicals into individual components. Once isolated, the components can be evaluated individually. In all chromatography, separation occurs when the sample mixture is introduced (injected) into a mobile phase. In liquid chromatography (LC), the mobile phase is a solvent. In gas chromatography (GC), the mobile phase is an inert gas such as helium. The mobile phase carries the sample mixture through what is referred to as a stationary phase. The stationary phase is usually a chemical that can selectively attract components in a sample mixture. The stationary phase is usually contained in a tube of some sort called a column. Columns can be glass or stainless steel of various dimensions. The mixture of compounds in the mobile phase interacts with the stationary phase. Each compound in the mixture interacts at a different rate. Those that interact the fastest will exit (elute from) the column first. Those that interact slowest will exit the column last. By changing characteristics of the mobile phase and the stationary phase, different mixtures of chemicals can be separated. -

Electrochemical Real-Time Mass Spectrometry: a Novel Tool for Time-Resolved Characterization of the Products of Electrochemical Reactions
Electrochemical real-time mass spectrometry: A novel tool for time-resolved characterization of the products of electrochemical reactions Elektrochemische Realzeit-Massenspektrometrie: Eine neuartige Methode zur zeitaufgelösten Charakterisierung der Produkte elektrochemischer Reaktionen Der Technischen Fakultät der Friedrich-Alexander-Universität Erlangen-Nürnberg zur Erlangung des Doktorgrades Dr.-Ingenieur vorgelegt von Peyman Khanipour Mehrin aus Shiraz, Iran Als Dissertation genehmigt von der Technischen Fakultät der Friedrich-Alexander-Universität Erlangen-Nürnberg Tag der mündlichen Prüfung: 17.11.2020 Vorsitzender des Promotionsorgans: Prof. Dr.-Ing. habil. Andreas Paul Fröba Gutachter: Prof. Dr. Karl J.J. Mayrhofer Prof. Dr. Frank-Michael Matysik I Acknowledgements This study is done in the electrosynthesis team of the electrocatalysis unit at Helmholtz- Institut Erlangen-Nürnberg (HI ERN) with the financial support of Forschungszentrum Jülich. I would like to express my deep gratitude to Prof. Dr. Karl J. J. Mayrhofer for accepting me as a Ph.D. student and also for all his encouragement, supports, and freedoms during my study. I’m grateful to Prof. Dr. Frank-Michael Matysik for kindly accepting to act as a second reviewer and also for the time he has invested in reading this thesis. This piece of work is enabled by collaboration with scientists from different expertise. I would like to express my appreciation to Dr. Sandra Haschke from FAU for providing shape-controlled high surface area platinum electrodes which I used for performing oxidation of primary alcohols and also the characterization of the provided material SEM, EDX, and XRD. Mr. Mario Löffler from HI ERN for obtaining the XPS data and his remarkable knowledge with the interpretation of the spectra on copper-based electrodes for the CO 2 electroreduction reaction. -

Ultra-Performance Liquid Chromatography Coupled to Quadrupole-Orthogonal Time-Of-flight Mass Spectrometry
RAPID COMMUNICATIONS IN MASS SPECTROMETRY Rapid Commun. Mass Spectrom. 2004; 18: 2331–2337 Published online in Wiley InterScience (www.interscience.wiley.com). DOI: 10.1002/rcm.1627 Ultra-performance liquid chromatography coupled to quadrupole-orthogonal time-of-flight mass spectrometry Robert Plumb1*, Jose Castro-Perez2, Jennifer Granger1, Iain Beattie3, Karine Joncour3 and Andrew Wright3 1Waters Corporation, Milford, MA, USA 2Waters Corporation, MS Technology Center, Manchester, UK 3AstraZeneca R&D Charnwood, Physical & Metabolic Science, Loughborough, UK Received 19 June 2004; Revised 5 August 2004; Accepted 9 August 2004 Ultra-performance liquid chromatography (UPLC) utilizes sub-2 mm particles with high linear sol- vent velocities to effect dramatic increases in resolution, sensitivity and speed of analysis. The reduction in particle size to below 2 mm requires instrumentation that can operate at pressures in the 6000–15 000 psi range. The typical peak widths generated by the UPLC system are in the order of 1–2 s for a 10-min separation. In the present work this technology has been applied to the study of in vivo drug metabolism, in particular the analysis of drug metabolites in bile. The reduction in peak width significantly increases analytical sensitivity by three- to five-fold, and the reduction in peak width, and concomitant increase in peak capacity, significantly reduces spectral overlap resulting in superior spectral quality in both MS and MS/MS modes. The application of UPLC/ MS resulted in the detection of additional drug metabolites, superior separation and improved spectral quality. Copyright # 2004 John Wiley & Sons, Ltd. The detection and identification of drug metabolites is crucial Liquid chromatography/mass spectrometry (LC/MS) and to both the drug discovery and development processes, LC/MS/MS have become the mainstay of the drug metabo- although in these two areas the emphasis is slightly different. -

Good Practice Guide for Isotope Ratio Mass Spectrometry, FIRMS (2011)
Good Practice Guide for Isotope Ratio Mass Spectrometry Good Practice Guide for Isotope Ratio Mass Spectrometry First Edition 2011 Editors Dr Jim Carter, UK Vicki Barwick, UK Contributors Dr Jim Carter, UK Dr Claire Lock, UK Acknowledgements Prof Wolfram Meier-Augenstein, UK This Guide has been produced by Dr Helen Kemp, UK members of the Steering Group of the Forensic Isotope Ratio Mass Dr Sabine Schneiders, Germany Spectrometry (FIRMS) Network. Dr Libby Stern, USA Acknowledgement of an individual does not indicate their agreement with Dr Gerard van der Peijl, Netherlands this Guide in its entirety. Production of this Guide was funded in part by the UK National Measurement System. This publication should be cited as: First edition 2011 J. F. Carter and V. J. Barwick (Eds), Good practice guide for isotope ratio mass spectrometry, FIRMS (2011). ISBN 978-0-948926-31-0 ISBN 978-0-948926-31-0 Copyright © 2011 Copyright of this document is vested in the members of the FIRMS Network. IRMS Guide 1st Ed. 2011 Preface A few decades ago, mass spectrometry (by which I mean organic MS) was considered a “black art”. Its complex and highly expensive instruments were maintained and operated by a few dedicated technicians and its output understood by only a few academics. Despite, or because, of this the data produced were amongst the “gold standard” of analytical science. In recent years a revolution occurred and MS became an affordable, easy to use and routine technique in many laboratories. Although many (rightly) applaud this popularisation, as a consequence the “black art” has been replaced by a “black box”: SAMPLES GO IN → → RESULTS COME OUT The user often has little comprehension of what goes on “under the hood” and, when “things go wrong”, the inexperienced operator can be unaware of why (or even that) the results that come out do not reflect the sample that goes in. -

Comparison of the Characteristic Mass Fragmentations of Phenethylamines and Tryptamines by Electron Ionization Gas Chromatograph
applied sciences Article Comparison of the Characteristic Mass Fragmentations of Phenethylamines and Tryptamines by Electron Ionization Gas Chromatography Mass Spectrometry, Electrospray and Matrix-Assisted Laser Desorption Ionization Mass Spectrometry Bo-Hong Chen, Ju-Tsung Liu, Hung-Ming Chen, Wen-Xiong Chen and Cheng-Huang Lin * Department of Chemistry, National Taiwan Normal University, 88 Sec. 4 Tingchow Road, Taipei 11677, Taiwan; [email protected] (B.-H.C.); [email protected] (J.-T.L.); [email protected] (H.-M.C.); [email protected] (W.-X.C.) * Correspondence: [email protected]; Tel.: +886-2-7734-6170; Fax: +886-2-2932-4249 Received: 18 April 2018; Accepted: 19 June 2018; Published: 22 June 2018 Abstract: Characteristic mass fragmentation of 20 phenethylamine/tryptamine standards were investigated and compared by means of matrix assisted laser desorption/time-of-flight mass spectrometry (MALDI/TOFM), gas chromatography–electron ionization–mass spectrometry (GC-EI/MS) and liquid chromatography–electrospray ionization/mass spectrometry (LC-ESI/MS) + methods. As a result, three characteristic peaks ([M] and fragments from the Cβ-Cα bond breakage) were found to be unique and contained information useful in identifying 2C series compounds based on the GC-EI/MS method. We found that the protonated molecular ion ([M+H]+) and two types of fragments produced from the α-cleavage and β-cleavage processes were useful mass spectral information in the rapid screening and confirmation of phenethylamine and tryptamine derivatives when ESI/MS and MALDI/TOFMS methods were applied. This assay was successfully used to determine samples that contain illicit drugs. Keywords: phenethylamine; tryptamine; MALDI/TOFMS; GC-EI/MS; LC-ESI/MS 1. -

Coupling Gas Chromatography to Mass Spectrometry
Coupling Gas Chromatography to Mass Spectrometry Introduction The suite of gas chromatographic detectors includes (roughly in order from most common to the least): the flame ionization detector (FID), thermal conductivity detector (TCD or hot wire detector), electron capture detector (ECD), photoionization detector (PID), flame photometric detector (FPD), thermionic detector, and a few more unusual or VERY expensive choices like the atomic emission detector (AED) and the ozone- or fluorine-induce chemiluminescence detectors. All of these except the AED produce an electrical signal that varies with the amount of analyte exiting the chromatographic column. The AED does that AND yields the emission spectrum of selected elements in the analytes as well. Another GC detector that is also very expensive but very powerful is a scaled down version of the mass spectrometer. When coupled to a GC the detection system itself is often referred to as the mass selective detector or more simply the mass detector. This powerful analytical technique belongs to the class of hyphenated analytical instrumentation (since each part had a different beginning and can exist independently) and is called gas chromatograhy/mass spectrometry (GC/MS). Placed at the end of a capillary column in a manner similar to the other GC detectors, the mass detector is more complicated than, for instance, the FID because of the mass spectrometer's complex requirements for the process of creation, separation, and detection of gas phase ions. A capillary column is required in the chromatograph because the entire MS process must be carried out at very low pressures (~10-5 torr) and in order to meet this requirement a vacuum is maintained via constant pumping using a vacuum pump. -

Electron Ionization
Chapter 6 Chapter 6 Electron Ionization I. Introduction ......................................................................................................317 II. Ionization Process............................................................................................317 III. Strategy for Data Interpretation......................................................................321 1. Assumptions 2. The Ionization Process IV. Types of Fragmentation Pathways.................................................................328 1. Sigma-Bond Cleavage 2. Homolytic or Radical-Site-Driven Cleavage 3. Heterolytic or Charge-Site-Driven Cleavage 4. Rearrangements A. Hydrogen-Shift Rearrangements B. Hydride-Shift Rearrangements V. Representative Fragmentations (Spectra) of Classes of Compounds.......... 344 1. Hydrocarbons A. Saturated Hydrocarbons 1) Straight-Chain Hydrocarbons 2) Branched Hydrocarbons 3) Cyclic Hydrocarbons B. Unsaturated C. Aromatic 2. Alkyl Halides 3. Oxygen-Containing Compounds A. Aliphatic Alcohols B. Aliphatic Ethers C. Aromatic Alcohols D. Cyclic Ethers E. Ketones and Aldehydes F. Aliphatic Acids and Esters G. Aromatic Acids and Esters 4. Nitrogen-Containing Compounds A. Aliphatic Amines B. Aromatic Compounds Containing Atoms of Nitrogen C. Heterocyclic Nitrogen-Containing Compounds D. Nitro Compounds E. Concluding Remarks on the Mass Spectra of Nitrogen-Containing Compounds 5. Multiple Heteroatoms or Heteroatoms and a Double Bond 6. Trimethylsilyl Derivative 7. Determining the Location of Double Bonds VI. Library -

Modern Mass Spectrometry
Modern Mass Spectrometry MacMillan Group Meeting 2005 Sandra Lee Key References: E. Uggerud, S. Petrie, D. K. Bohme, F. Turecek, D. Schröder, H. Schwarz, D. Plattner, T. Wyttenbach, M. T. Bowers, P. B. Armentrout, S. A. Truger, T. Junker, G. Suizdak, Mark Brönstrup. Topics in Current Chemistry: Modern Mass Spectroscopy, pp. 1-302, 225. Springer-Verlag, Berlin, 2003. Current Topics in Organic Chemistry 2003, 15, 1503-1624 1 The Basics of Mass Spectroscopy ! Purpose Mass spectrometers use the difference in mass-to-charge ratio (m/z) of ionized atoms or molecules to separate them. Therefore, mass spectroscopy allows quantitation of atoms or molecules and provides structural information by the identification of distinctive fragmentation patterns. The general operation of a mass spectrometer is: "1. " create gas-phase ions "2. " separate the ions in space or time based on their mass-to-charge ratio "3. " measure the quantity of ions of each mass-to-charge ratio Ionization sources ! Instrumentation Chemical Ionisation (CI) Atmospheric Pressure CI!(APCI) Electron Impact!(EI) Electrospray Ionization!(ESI) SORTING DETECTION IONIZATION OF IONS OF IONS Fast Atom Bombardment (FAB) Field Desorption/Field Ionisation (FD/FI) Matrix Assisted Laser Desorption gaseous mass ion Ionisation!(MALDI) ion source analyzer transducer Thermospray Ionisation (TI) Analyzers quadrupoles vacuum signal Time-of-Flight (TOF) pump processor magnetic sectors 10-5– 10-8 torr Fourier transform and quadrupole ion traps inlet Detectors mass electron multiplier spectrum Faraday cup Ionization Sources: Classical Methods ! Electron Impact Ionization A beam of electrons passes through a gas-phase sample and collides with neutral analyte molcules (M) to produce a positively charged ion or a fragment ion. -

Find the Molar Mass of Sodium Carbonate, Na 2CO3. Na 2 X
Moles and Molar Mass The mole is the "counting unit" used by chemists to indicate the number of atoms, ions, molecules, or formula units present in a particular chemical sample. The mole is similar to other counting units that you've used before....pair (2), dozen (12), and gross (144). One mole of a compound contains Avogadro's number (6.022 x 1023) of molecules (molecular compound) or formula units (ionic compound). The molar mass of a compound tells you the mass of 1 mole of that substance. In other words, it tells you the number of grams per mole of a compound. The units for molar mass are, therefore, grams/mole. To find the molar mass of a compound: 1. Use the chemical formula to determine the number of each type of atom present in the compound. 2. Multiply the atomic weight (from the periodic table) of each element by the number of atoms of that element present in the compound. 3. Add it all together and put units of grams/mole after the number. Example: Find the molar mass of sodium carbonate, Na2CO3. Na 2 x 23.0 = 46.0 C 1 x 12.0 = 12.0 O 3 x 16.0 = 48.0 molar = 106.0 g/mole mass For many (but not all) problems, you can simply round the atomic weights and the molar mass to the nearest 0.1 g/mole. HOWEVER, make sure that you use at least as many significant figures in your molar mass as the measurement with the fewest significant figures. -
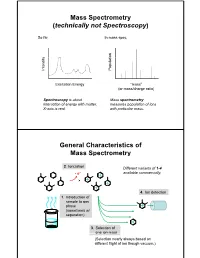
Mass Spectrometry (Technically Not Spectroscopy)
Mass Spectrometry (technically not Spectroscopy) So far, In mass spec, on y Populati Intensit Excitation Energy “mass” (or mass/charge ratio) Spectroscopy is about Mass spectrometry interaction of energy with matter. measures population of ions X-axis is real. with particular mass. General Characteristics of Mass Spectrometry 2. Ionization Different variants of 1-4 -e- available commercially. 4. Ion detection 1. Introduction of sample to gas phase (sometimes w/ separation) 3. Selection of one ion mass (Selection nearly always based on different flight of ion though vacuum.) General Components of a Mass Spectrometer Lots of choices, which can be mixed and matched. direct injection The Mass Spectrum fragment “daughter” ions M+ “parent” mass Sample Introduction: Direc t Inser tion Prob e If sample is a liquid, sample can also be injected directly into ionization region. If sample isn’t pure, get multiple parents (that can’t be distinguished from fragments). Capillary Column Introduction Continous source of molecules to spectrometer. detector column (including GC, LC, chiral, size exclusion) • Signal intensity depends on both amount of molecule and ionization efficiency • To use quantitatively, must calibrate peaks with respect eltilution time ttlitotal ion curren t to quantity eluted (TIC) over time Capillary Column Introduction Easy to interface with gas or liquid chromatography. TIC trace elution time time averaged time averaged mass spectrum mass spectrum Methods of Ionization: Electron Ionization (EI) 1 - + - 1 M + e (kV energy) M + 2e Fragmentation in Electron Ionization daughter ion (observed in spectrum) neutral fragment (not observed) excited parent at electron at electron energy of energy of 15 eV 70 e V Lower electron energy yields less fragmentation, but also less signal.