Ultra-Performance Liquid Chromatography Coupled to Quadrupole-Orthogonal Time-Of-flight Mass Spectrometry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gas Chromatography-Mass Spectroscopy
Gas Chromatography-Mass Spectroscopy Introduction Gas chromatography-mass spectroscopy (GC-MS) is one of the so-called hyphenated analytical techniques. As the name implies, it is actually two techniques that are combined to form a single method of analyzing mixtures of chemicals. Gas chromatography separates the components of a mixture and mass spectroscopy characterizes each of the components individually. By combining the two techniques, an analytical chemist can both qualitatively and quantitatively evaluate a solution containing a number of chemicals. Gas Chromatography In general, chromatography is used to separate mixtures of chemicals into individual components. Once isolated, the components can be evaluated individually. In all chromatography, separation occurs when the sample mixture is introduced (injected) into a mobile phase. In liquid chromatography (LC), the mobile phase is a solvent. In gas chromatography (GC), the mobile phase is an inert gas such as helium. The mobile phase carries the sample mixture through what is referred to as a stationary phase. The stationary phase is usually a chemical that can selectively attract components in a sample mixture. The stationary phase is usually contained in a tube of some sort called a column. Columns can be glass or stainless steel of various dimensions. The mixture of compounds in the mobile phase interacts with the stationary phase. Each compound in the mixture interacts at a different rate. Those that interact the fastest will exit (elute from) the column first. Those that interact slowest will exit the column last. By changing characteristics of the mobile phase and the stationary phase, different mixtures of chemicals can be separated. -

Electrochemical Real-Time Mass Spectrometry: a Novel Tool for Time-Resolved Characterization of the Products of Electrochemical Reactions
Electrochemical real-time mass spectrometry: A novel tool for time-resolved characterization of the products of electrochemical reactions Elektrochemische Realzeit-Massenspektrometrie: Eine neuartige Methode zur zeitaufgelösten Charakterisierung der Produkte elektrochemischer Reaktionen Der Technischen Fakultät der Friedrich-Alexander-Universität Erlangen-Nürnberg zur Erlangung des Doktorgrades Dr.-Ingenieur vorgelegt von Peyman Khanipour Mehrin aus Shiraz, Iran Als Dissertation genehmigt von der Technischen Fakultät der Friedrich-Alexander-Universität Erlangen-Nürnberg Tag der mündlichen Prüfung: 17.11.2020 Vorsitzender des Promotionsorgans: Prof. Dr.-Ing. habil. Andreas Paul Fröba Gutachter: Prof. Dr. Karl J.J. Mayrhofer Prof. Dr. Frank-Michael Matysik I Acknowledgements This study is done in the electrosynthesis team of the electrocatalysis unit at Helmholtz- Institut Erlangen-Nürnberg (HI ERN) with the financial support of Forschungszentrum Jülich. I would like to express my deep gratitude to Prof. Dr. Karl J. J. Mayrhofer for accepting me as a Ph.D. student and also for all his encouragement, supports, and freedoms during my study. I’m grateful to Prof. Dr. Frank-Michael Matysik for kindly accepting to act as a second reviewer and also for the time he has invested in reading this thesis. This piece of work is enabled by collaboration with scientists from different expertise. I would like to express my appreciation to Dr. Sandra Haschke from FAU for providing shape-controlled high surface area platinum electrodes which I used for performing oxidation of primary alcohols and also the characterization of the provided material SEM, EDX, and XRD. Mr. Mario Löffler from HI ERN for obtaining the XPS data and his remarkable knowledge with the interpretation of the spectra on copper-based electrodes for the CO 2 electroreduction reaction. -

Coupling Gas Chromatography to Mass Spectrometry
Coupling Gas Chromatography to Mass Spectrometry Introduction The suite of gas chromatographic detectors includes (roughly in order from most common to the least): the flame ionization detector (FID), thermal conductivity detector (TCD or hot wire detector), electron capture detector (ECD), photoionization detector (PID), flame photometric detector (FPD), thermionic detector, and a few more unusual or VERY expensive choices like the atomic emission detector (AED) and the ozone- or fluorine-induce chemiluminescence detectors. All of these except the AED produce an electrical signal that varies with the amount of analyte exiting the chromatographic column. The AED does that AND yields the emission spectrum of selected elements in the analytes as well. Another GC detector that is also very expensive but very powerful is a scaled down version of the mass spectrometer. When coupled to a GC the detection system itself is often referred to as the mass selective detector or more simply the mass detector. This powerful analytical technique belongs to the class of hyphenated analytical instrumentation (since each part had a different beginning and can exist independently) and is called gas chromatograhy/mass spectrometry (GC/MS). Placed at the end of a capillary column in a manner similar to the other GC detectors, the mass detector is more complicated than, for instance, the FID because of the mass spectrometer's complex requirements for the process of creation, separation, and detection of gas phase ions. A capillary column is required in the chromatograph because the entire MS process must be carried out at very low pressures (~10-5 torr) and in order to meet this requirement a vacuum is maintained via constant pumping using a vacuum pump. -

Electron Ionization
Chapter 6 Chapter 6 Electron Ionization I. Introduction ......................................................................................................317 II. Ionization Process............................................................................................317 III. Strategy for Data Interpretation......................................................................321 1. Assumptions 2. The Ionization Process IV. Types of Fragmentation Pathways.................................................................328 1. Sigma-Bond Cleavage 2. Homolytic or Radical-Site-Driven Cleavage 3. Heterolytic or Charge-Site-Driven Cleavage 4. Rearrangements A. Hydrogen-Shift Rearrangements B. Hydride-Shift Rearrangements V. Representative Fragmentations (Spectra) of Classes of Compounds.......... 344 1. Hydrocarbons A. Saturated Hydrocarbons 1) Straight-Chain Hydrocarbons 2) Branched Hydrocarbons 3) Cyclic Hydrocarbons B. Unsaturated C. Aromatic 2. Alkyl Halides 3. Oxygen-Containing Compounds A. Aliphatic Alcohols B. Aliphatic Ethers C. Aromatic Alcohols D. Cyclic Ethers E. Ketones and Aldehydes F. Aliphatic Acids and Esters G. Aromatic Acids and Esters 4. Nitrogen-Containing Compounds A. Aliphatic Amines B. Aromatic Compounds Containing Atoms of Nitrogen C. Heterocyclic Nitrogen-Containing Compounds D. Nitro Compounds E. Concluding Remarks on the Mass Spectra of Nitrogen-Containing Compounds 5. Multiple Heteroatoms or Heteroatoms and a Double Bond 6. Trimethylsilyl Derivative 7. Determining the Location of Double Bonds VI. Library -

Fundamentals of Biological Mass Spectrometry and Proteomics
Fundamentals of Biological Mass Spectrometry and Proteomics Steve Carr Broad Institute of MIT and Harvard Modern Mass Spectrometer (MS) Systems Orbitrap Q-Exactive Triple Quadrupole Discovery/Global Experiments Targeted MS MS systems used for proteomics have 4 tasks: • Create ions from analyte molecules • Separate the ions based on charge and mass • Detect ions and determine their mass-to-charge • Select and fragment ions of interest to provide structural information (MS/MS) Electrospray MS: ease of coupling to liquid-based separation methods has made it the key technology in proteomics Possible Sample Inlets Syringe Pump Sample Injection Loop Liquid Autosampler, HPLC Capillary Electrophoresis Expansion of the Ion Formation and Sampling Regions Nitrogen Drying Gas Electrospray Atmosphere Vacuum Needle 3- 5 kV Liquid Nebulizing Gas Droplets Ions Containing Solvated Ions Isotopes Most elements have more than one stable isotope. For example, most carbon atoms have a mass of 12 Da, but in nature, 1.1% of C atoms have an extra neutron, making their mass 13 Da. Why do we care? Mass spectrometers “see” the isotope peaks provided the resolution is high enough. If an MS instrument has resolution high enough to resolve these isotopes, better mass accuracy is achieved. Stable isotopes of most abundant elements of peptides Element Mass Abundance H 1.0078 99.985% 2.0141 0.015 C 12.0000 98.89 13.0034 1.11 N 14.0031 99.64 15.0001 0.36 O 15.9949 99.76 16.9991 0.04 17.9992 0.20 Monoisotopic mass and isotopes We use instruments that resolve the isotopes enabling us to accurately measure the monoisotopic mass MonoisotopicMonoisotopic mass; all 12C, mass no 13C atoms corresponds to 13 lowestOne massC atom peak Two 13C atoms Angiotensin I (MW = 1295.6) (M+H)+ = C62 H90 N17 O14 TheWhen monoisotopic the isotopes mass of aare molecule clearly is the resolved sum of the the accurate monoisotopic masses for the massmost abundant isotope of each element present. -
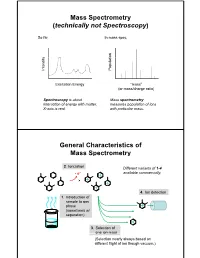
Mass Spectrometry (Technically Not Spectroscopy)
Mass Spectrometry (technically not Spectroscopy) So far, In mass spec, on y Populati Intensit Excitation Energy “mass” (or mass/charge ratio) Spectroscopy is about Mass spectrometry interaction of energy with matter. measures population of ions X-axis is real. with particular mass. General Characteristics of Mass Spectrometry 2. Ionization Different variants of 1-4 -e- available commercially. 4. Ion detection 1. Introduction of sample to gas phase (sometimes w/ separation) 3. Selection of one ion mass (Selection nearly always based on different flight of ion though vacuum.) General Components of a Mass Spectrometer Lots of choices, which can be mixed and matched. direct injection The Mass Spectrum fragment “daughter” ions M+ “parent” mass Sample Introduction: Direc t Inser tion Prob e If sample is a liquid, sample can also be injected directly into ionization region. If sample isn’t pure, get multiple parents (that can’t be distinguished from fragments). Capillary Column Introduction Continous source of molecules to spectrometer. detector column (including GC, LC, chiral, size exclusion) • Signal intensity depends on both amount of molecule and ionization efficiency • To use quantitatively, must calibrate peaks with respect eltilution time ttlitotal ion curren t to quantity eluted (TIC) over time Capillary Column Introduction Easy to interface with gas or liquid chromatography. TIC trace elution time time averaged time averaged mass spectrum mass spectrum Methods of Ionization: Electron Ionization (EI) 1 - + - 1 M + e (kV energy) M + 2e Fragmentation in Electron Ionization daughter ion (observed in spectrum) neutral fragment (not observed) excited parent at electron at electron energy of energy of 15 eV 70 e V Lower electron energy yields less fragmentation, but also less signal. -

Cryogenic Micro-Calorimeters for Mass Spectrometry of Kev Neutral Atoms
Journal of Physics: Conference Series PAPER • OPEN ACCESS Related content - The status of the micro-calorimeter at the Cryogenic micro-calorimeters for mass Shanghai EBIT - The Variability of the Recombination of spectrometry of keV neutral atoms and molecules Hydrogen Atoms on Metal Surfaces - SQUID gradiometer for ultra-low To cite this article: O Novotný et al 2015 J. Phys.: Conf. Ser. 635 032023 temperature magnetic micro-calorimeter View the article online for updates and enhancements. This content was downloaded from IP address 129.13.72.197 on 04/11/2020 at 09:19 XXIX International Conference on Photonic, Electronic, and Atomic Collisions (ICPEAC2015) IOP Publishing Journal of Physics: Conference Series 635 (2015) 032023 doi:10.1088/1742-6596/635/3/032023 Cryogenic micro-calorimeters for mass spectrometry of keV neutral atoms and molecules O Novotn´y∗;y 1, S Allgeierz, C Enssz, A Fleischmannz, L Gamerz, D Hengstlerz, S Kempfz, C Krantz∗, A Pabingerz, C Piesz, D W Saviny, D Schwalm∗;⋄, A Wolf∗ 2 ∗ Max Planck Institute for Nuclear Physics, D-69117 Heidelberg, Germany y Columbia Astrophysics Laboratory, Columbia University, New York, NY 10027, USA z Kirchhoff Institute for Physics, Heidelberg University, D-69120 Heidelberg, Germany ⋄ Faculty of Physics, Weizmann Institute of Science, Rehovot 76100, Israel Synopsis We demonstrate the capability of micro-calorimeters to detect and mass-resolve neutral atoms and molecules at ∼ keV energies, reaching single H-atom resolution. Mass spectrometric identification of fragmen- also studied in detail the MMC response function tation products is one of the most important for molecular projectiles of masses up to 56 amu. -

An Organic Chemist's Guide to Electrospray Mass Spectrometric
molecules Review An Organic Chemist’s Guide to Electrospray Mass Spectrometric Structure Elucidation Arnold Steckel 1 and Gitta Schlosser 2,* 1 Hevesy György PhD School of Chemistry, ELTE Eötvös Loránd University, Pázmány Péter sétány 1/A, 1117 Budapest, Hungary; [email protected] 2 Department of Analytical Chemistry, ELTE Eötvös Loránd University, Pázmány Péter sétány 1/A, 1117 Budapest, Hungary * Correspondence: [email protected] Received: 16 January 2019; Accepted: 8 February 2019; Published: 10 February 2019 Abstract: Tandem mass spectrometry is an important tool for structure elucidation of natural and synthetic organic products. Fragmentation of odd electron ions (OE+) generated by electron ionization (EI) was extensively studied in the last few decades, however there are only a few systematic reviews available concerning the fragmentation of even-electron ions (EE+/EE−) produced by the currently most common ionization techniques, electrospray ionization (ESI) and atmospheric pressure chemical ionization (APCI). This review summarizes the most important features of tandem mass spectra generated by collision-induced dissociation fragmentation and presents didactic examples for the unexperienced users. Keywords: tandem mass spectrometry; MS/MS fragmentation; collision-induced dissociation; CID; ESI; structure elucidation 1. Introduction Electron ionization (EI), a hard ionization technique, is the method of choice for analyses of small (<1000 Da), nonpolar, volatile compounds. As its name implies, the technique involves ionization by electrons with ~70 eV energy. This energy is high enough to yield very reproducible mass spectra with a large number of fragments. However, these spectra frequently lack the radical type molecular ions (M+) due to the high internal energy transferred to the precursors [1]. -

Electrochemistry As an Adjunct to Mass Spectrometry in Drug Development
Application Note Application Electrochemistry as an Adjunct to Mass Spectrometry in Drug Development Ian N. Acworth, David Thomas, Paul Gamache Thermo Fisher Scientific, Chelmsford, MA 1016 Key Words Drug bioactivation, Stability and degradation, Electrochemical detection, Enhanced ionization, Drug metabolites, Reactive intermediate Goal To show how on-line HPLC-electrochemical oxidation-mass spectrometry can be harnessed to accelerate several key steps in drug development. Introduction Oxidation plays a number of critical roles in biology and drug metabolism. In addition to being the major process for generating energy in a living cell, it is the major mechanism by which drugs are metabolized. Oxidation is a contributor to drug bioactivation and idiosyncratic Experimental toxicity, and is also involved in drug stability and Applications were developed using gradient analytical or degradation. Electrochemistry (EC) when used with HPLC semi-preparative HPLC with pre- or post-column has long been recognized as a sensitive and selective electrochemical oxidation followed by mass spectrometric detection technique. Indeed more than 90% of detection. Drug stability studies used flow injection pharmaceuticals in the marketplace can be oxidized and analysis with coulometric array detection. detected using this technique. However, the inherent Sample Preparation electrochemical nature of many drugs goes beyond simple Samples were dissolved in initial mobile phase solution to analysis. This commonality of oxidation makes EC a concentration of 20 -

Calorimetry Has Been Routinely Used at US and European Facilities For
10. PRINCIPLES AND APPLICATIONS OF CALORIMETRIC ASSAY D. S. Bracken, and C. R. Rudy I. Introduction Calorimetry is the quantitative measurement of heat. Applications of calorimetry include measurements of the specific heats of elements and compounds, phase-change enthalpies, and the rate of heat generation from radionuclides. The most successful radiometric calorimeter designs fit the general category of heat-flow calorimeters. Calorimetry is used as a nondestructive assay (NDA) technique for determining the power output of heat-producing nuclear materials. The heat is generated by the decay of radioactive isotopes within the item. Because the heat-measurement result is completely independent of material and matrix type, it can be used on any material form or item matrix. Heat-flow calorimeters have been used to measure thermal powers from 0.5 mW (0.2 g low-burnup plutonium equivalent) to 1,000 W for items ranging in size from less than 2.54 cm to 60 cm in diameter and up to 100 cm in length. Calorimetric assay is the determination of the mass of radioactive material through the combined measurement of its thermal power by calorimetry and its isotopic composition by gamma-ray spectroscopy or mass spectroscopy. Calorimetric assay has been routinely used at U.S. and European facilities for plutonium process measurements and nuclear material accountability for the last 40 years [EI54, GU64, GU70, ANN15.22, AS1458, MA82, IAEA87]. Calorimetric assay is routinely used as a reliable NDA technique for the quantification of plutonium and tritium content. Calorimetric assay of tritium and plutonium-bearing items routinely obtains the highest precision and accuracy of all NDA techniques. -

LILBID Laser Dissociation Curves: a Mass Spectrometry-Based Method
www.nature.com/scientificreports OPEN LILBID laser dissociation curves: a mass spectrometry‑based method for the quantitative assessment of dsDNA binding afnities Phoebe Young1, Genia Hense1, Carina Immer2,3, Jens Wöhnert2,3 & Nina Morgner1* One current goal in native mass spectrometry is the assignment of binding afnities to noncovalent complexes. Here we introduce a novel implementation of the existing laser‑induced liquid bead ion desorption (LILBID) mass spectrometry method: this new method, LILBID laser dissociation curves, assesses binding strengths quantitatively. In all LILBID applications, aqueous sample droplets are irradiated by 3 µm laser pulses. Variation of the laser energy transferred to the droplet during desorption afects the degree of complex dissociation. In LILBID laser dissociation curves, laser energy transfer is purposely varied, and a binding afnity is calculated from the resulting complex dissociation. A series of dsDNAs with diferent binding afnities was assessed using LILBID laser dissociation curves. The binding afnity results from the LILBID laser dissociation curves strongly correlated with the melting temperatures from UV melting curves and with dissociation constants from isothermal titration calorimetry, standard solution phase methods. LILBID laser dissociation curve data also showed good reproducibility and successfully predicted the melting temperatures and dissociation constants of three DNA sequences. LILBID laser dissociation curves are a promising native mass spectrometry binding afnity method, with reduced time and sample consumption compared to melting curves or titrations. Native mass spectrometry (MS) was introduced in the 1990s as a method to study intact noncovalent biomolecu- lar complexes1,2, focusing primarily on their stoichiometry. However, to understand the function of such com- plexes, it is important not only to know their composition but also to characterize the binding afnities of their components. -

Print Special Issue Flyer
IMPACT FACTOR 3.623 an Open Access Journal by MDPI Mass Spectrometry in Materials Science Guest Editor: Message from the Guest Editor Dr. Rafał Frański Dear colleagues, Adam Mickiewicz University, Faculty of Chemistry, Poznań, Mass spectrometry (MS) has become an important tool for Poland scientists working on the development of modern [email protected] materials. The main mass spectrometry techniques which have advanced our knowledge in the field of material science are secondary ion mass spectrometry (SIMS), Deadline for manuscript inductively coupled plasma mass spectrometry (ICP-MS), submissions: and laser ablation inductively coupled plasma mass 10 May 2022 spectrometry (LA-ICP-MS). This Special Issue of Materials, “Mass Spectrometry in Materials Science”, will focus on the application of mass spectrometry to the ultra-trace analysis, micro and nanodistribution analysis, surface analysis, as well as three-dimensional analysis of different kinds of advanced materials, e.g., semiconductors, superconductors, glass, metals and their oxides, biomaterials, ceramic materials, stainless steels, and others. Authors are invited to submit manuscripts that use mass spectrometry as an important tool in high-resolution material analysis and characterization. Dr. Rafał Frański Guest Editor mdpi.com/si/43630 SpeciaIslsue IMPACT FACTOR 3.623 an Open Access Journal by MDPI Editor-in-Chief Message from the Editor-in-Chief Prof. Dr. Maryam Tabrizian Materials (ISSN 1996-1944) was launched in 2008. The James McGill Professor, journal covers twenty comprehensive