Advanced Separation Techniques Combined with Mass Spectrometry for Difficult Analytical Tasks - Isomer Separation and Oil Analysis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gas Chromatography-Mass Spectroscopy
Gas Chromatography-Mass Spectroscopy Introduction Gas chromatography-mass spectroscopy (GC-MS) is one of the so-called hyphenated analytical techniques. As the name implies, it is actually two techniques that are combined to form a single method of analyzing mixtures of chemicals. Gas chromatography separates the components of a mixture and mass spectroscopy characterizes each of the components individually. By combining the two techniques, an analytical chemist can both qualitatively and quantitatively evaluate a solution containing a number of chemicals. Gas Chromatography In general, chromatography is used to separate mixtures of chemicals into individual components. Once isolated, the components can be evaluated individually. In all chromatography, separation occurs when the sample mixture is introduced (injected) into a mobile phase. In liquid chromatography (LC), the mobile phase is a solvent. In gas chromatography (GC), the mobile phase is an inert gas such as helium. The mobile phase carries the sample mixture through what is referred to as a stationary phase. The stationary phase is usually a chemical that can selectively attract components in a sample mixture. The stationary phase is usually contained in a tube of some sort called a column. Columns can be glass or stainless steel of various dimensions. The mixture of compounds in the mobile phase interacts with the stationary phase. Each compound in the mixture interacts at a different rate. Those that interact the fastest will exit (elute from) the column first. Those that interact slowest will exit the column last. By changing characteristics of the mobile phase and the stationary phase, different mixtures of chemicals can be separated. -

Section 13. Disposal Considerations Disposal Methods : the Generation of Waste Should Be Avoided Or Minimized Wherever Possible
SAFETY DATA SHEET Flammable Liquid Mixture: Docosane / Dodecane / Dotriacontane / Eicosane / Heptane / Hexacosane / Hexadecane / Hexane / N-Decane / N-Nonane / N- Octadecane / Octacosane / Octane / Tetracosane / Tetradecane / Triacontane Section 1. Identification GHS product identifier : Flammable Liquid Mixture: Docosane / Dodecane / Dotriacontane / Eicosane / Heptane / Hexacosane / Hexadecane / Hexane / N-Decane / N-Nonane / N-Octadecane / Octacosane / Octane / Tetracosane / Tetradecane / Triacontane Other means of : Not available. identification Product use : Synthetic/Analytical chemistry. SDS # : 019981 Supplier's details : Airgas USA, LLC and its affiliates 259 North Radnor-Chester Road Suite 100 Radnor, PA 19087-5283 1-610-687-5253 24-hour telephone : 1-866-734-3438 Section 2. Hazards identification OSHA/HCS status : This material is considered hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200). Classification of the : FLAMMABLE LIQUIDS - Category 1 substance or mixture SKIN CORROSION/IRRITATION - Category 2 SERIOUS EYE DAMAGE/ EYE IRRITATION - Category 2A TOXIC TO REPRODUCTION (Fertility) - Category 2 TOXIC TO REPRODUCTION (Unborn child) - Category 2 SPECIFIC TARGET ORGAN TOXICITY (SINGLE EXPOSURE) (Narcotic effects) - Category 3 SPECIFIC TARGET ORGAN TOXICITY (REPEATED EXPOSURE) - Category 2 AQUATIC HAZARD (ACUTE) - Category 2 AQUATIC HAZARD (LONG-TERM) - Category 1 GHS label elements Hazard pictograms : Signal word : Danger Hazard statements : Extremely flammable liquid and vapor. May form explosive mixtures in Air. Causes serious eye irritation. Causes skin irritation. Suspected of damaging fertility or the unborn child. May cause drowsiness and dizziness. May cause damage to organs through prolonged or repeated exposure. Very toxic to aquatic life with long lasting effects. Precautionary statements General : Read label before use. Keep out of reach of children. If medical advice is needed, have product container or label at hand. -

Qualitative and Quantitative Analysis
qualitative and quantitative analysis Russian scientist Tswett in 1906 used a glass columns packed with divided CaCO3(calcium carbonate) to separate plant pigments extracted by hexane. The pigments after separation appeared as colour bands that can come out of the column one by one. Tswett was the first to use the term "chromatography" derived from two Greek words "Chroma" meaning color and "graphein" meaning to write. Invention of Chromatography by M. Tswett Ether Chromatography Colors Chlorophyll CaCO3 5 *Definition of chromatography *Tswett (1906) stated that „chromatography is a method in which the components of a mixture are separated on adsorbent column in a flowing system”. *IUPAC definition (International Union of pure and applied Chemistry) (1993): Chromatography is a physical method of separation in which the components to be separated are distributed between two phases, one of which is stationary while the other moves in a definite direction. *Principles of Chromatography * Any chromatography system is composed of three components : * Stationary phase * Mobile phase * Mixture to be separated The separation process occurs because the components of mixture have different affinities for the two phases and thus move through the system at different rates. A component with a high affinity for the mobile phase moves quickly through the chromatographic system, whereas one with high affinity for the solid phase moves more slowly. *Forces Responsible for Separation * The affinity differences of the components for the stationary or the mobile phases can be due to several different chemical or physical properties including: * Ionization state * Polarity and polarizability * Hydrogen bonding / van der Waals’ forces * Hydrophobicity * Hydrophilicity * The rate at which a sample moves is determined by how much time it spends in the mobile phase. -

Synthetic Turf Scientific Advisory Panel Meeting Materials
California Environmental Protection Agency Office of Environmental Health Hazard Assessment Synthetic Turf Study Synthetic Turf Scientific Advisory Panel Meeting May 31, 2019 MEETING MATERIALS THIS PAGE LEFT BLANK INTENTIONALLY Office of Environmental Health Hazard Assessment California Environmental Protection Agency Agenda Synthetic Turf Scientific Advisory Panel Meeting May 31, 2019, 9:30 a.m. – 4:00 p.m. 1001 I Street, CalEPA Headquarters Building, Sacramento Byron Sher Auditorium The agenda for this meeting is given below. The order of items on the agenda is provided for general reference only. The order in which items are taken up by the Panel is subject to change. 1. Welcome and Opening Remarks 2. Synthetic Turf and Playground Studies Overview 4. Synthetic Turf Field Exposure Model Exposure Equations Exposure Parameters 3. Non-Targeted Chemical Analysis Volatile Organics on Synthetic Turf Fields Non-Polar Organics Constituents in Crumb Rubber Polar Organic Constituents in Crumb Rubber 5. Public Comments: For members of the public attending in-person: Comments will be limited to three minutes per commenter. For members of the public attending via the internet: Comments may be sent via email to [email protected]. Email comments will be read aloud, up to three minutes each, by staff of OEHHA during the public comment period, as time allows. 6. Further Panel Discussion and Closing Remarks 7. Wrap Up and Adjournment Agenda Synthetic Turf Advisory Panel Meeting May 31, 2019 THIS PAGE LEFT BLANK INTENTIONALLY Office of Environmental Health Hazard Assessment California Environmental Protection Agency DRAFT for Discussion at May 2019 SAP Meeting. Table of Contents Synthetic Turf and Playground Studies Overview May 2019 Update ..... -

Sampling Dynamics for Volatile Organic Compounds Using Headspace Solid-Phase Microextraction Arrow for Microbiological Samples
separations Communication Sampling Dynamics for Volatile Organic Compounds Using Headspace Solid-Phase Microextraction Arrow for Microbiological Samples Kevin E. Eckert 1, David O. Carter 2 and Katelynn A. Perrault 1,* 1 Laboratory of Forensic and Bioanalytical Chemistry, Forensic Sciences Unit, Division of Natural Sciences and Mathematics, Chaminade University of Honolulu, 3140 Waialae Ave., Honolulu, HI 96816, USA; [email protected] 2 Laboratory of Forensic Taphonomy, Forensic Sciences Unit, Division of Natural Sciences and Mathematics, Chaminade University of Honolulu, 3140 Waialae Avenue, Honolulu, HI 96816, USA; [email protected] * Correspondence: [email protected] Received: 10 August 2018; Accepted: 27 August 2018; Published: 10 September 2018 Abstract: Volatile organic compounds (VOCs) are monitored in numerous fields using several commercially-available sampling options. Sorbent-based sampling techniques, such as solid-phase microextraction (SPME), provide pre-concentration and focusing of VOCs prior to gas chromatography–mass spectrometry (GC–MS) analysis. This study investigated the dynamics of SPME Arrow, which exhibits an increased sorbent phase volume and improved durability compared to traditional SPME fibers. A volatile reference mixture (VRM) and saturated alkanes mix (SAM) were used to investigate optimal parameters for microbiological VOC profiling in combination with GC–MS analysis. Fiber type, extraction time, desorption time, carryover, and reproducibility were characterized, in addition to a comparison with traditional SPME fibers. The developed method was then applied to longitudinal monitoring of Bacillus subtilis cultures, which represents a ubiquitous microbe in medical, forensic, and agricultural applications. The carbon wide range/polydimethylsiloxane (CWR/PDMS) fiber was found to be optimal for the range of expected VOCs in microbiological profiling, and a statistically significant increase in the majority of VOCs monitored was observed. -

Laser Desorption/Ionization Time-Of-Flight Mass Spectrometry of Biomolecules Yu-Chen Chang Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1996 Laser desorption/ionization time-of-flight mass spectrometry of biomolecules Yu-chen Chang Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Analytical Chemistry Commons Recommended Citation Chang, Yu-chen, "Laser desorption/ionization time-of-flight mass spectrometry of biomolecules " (1996). Retrospective Theses and Dissertations. 11366. https://lib.dr.iastate.edu/rtd/11366 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. INFORMATION TO USERS This manuscript has been rqjroduced fix>m the microfihn master. UMI fihns the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, w^e others may be from any type of computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely afifect reproduction. In the unlikely event that the author did not send IMl a complete manuscript and there are misang pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. Oversize materials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand comer and continuing from left to right in equal sections with small overlaps. -

Separations Development and Application (WBS 2.4.1.101) U.S
Separations Development and Application (WBS 2.4.1.101) U.S. Department of Energy (DOE) Bioenergy Technologies Office (BETO) 2017 Project Peer Review James D. (“Jim”) McMillan NREL March 7, 2017 Biochemical Conversion Session This presentation does not contain any proprietary, confidential, or otherwise restricted information. Project Goal Overall goal: Define, develop and apply separation processes to enable cost- effective hydrocarbon fuel / precursor production; focus on sugars and fuel precursor streams; lipids pathway shown. Outcome: Down selected proven, viable methods for clarifying and concentrating the sugar intermediates stream and for recovering lipids from oleaginous yeast that pass “go” criteria (i.e., high yield, scalable, and cost effective). Relevance: Separations are key to overall process integration and economics; often represent ≥ 50% of total process costs; performance/efficiency can make or break process viability. Separations this project investigates – sugar stream clarification and concentration, and recovery of intracellular lipids from yeast – account for 17-26% of projected Minimum Fuel Selling Price (MFSP) for the integrated process. 2 Project Overview • Cost driven R&D to assess/develop/improve key process separations - Sugar stream separations: S/L, concentrative and polishing - Fuel precursor recovery separations: oleaginous yeast cell lysis and LLE lipid recovery • Identify and characterize effective methods - Show capability to pass relevant go/no-go criteria (e.g., high yield, low cost, scalable) • Exploit in situ separation for process intensification - Enable Continuous Enzymatic Hydrolysis (CEH) • Generate performance data to develop / refine process TEAs and LCAs 3 Separations Technoeconomic Impact $1.78 $2.18 $1.43 $0.79 4 Quad Chart Overview Timeline Barriers Primary focus on addressing upstream and Start: FY 15 (Oct., ‘14) downstream separations-related barriers: End: FY 17 (Sept., ‘17; projected) – Ct-G. -

Rubber-Modified Epoxies: in Situ Detection of the Phase Separation by Differential Scanning Calorimetry
Rubber-modified epoxies: in situ detection of the phase separation by differential scanning calorimetry Dong Pu Fang*, C. C. Riccardi and R. J. J. Williamst Institute of Materials Science and Technology (INTEMA), University of Mar de/Plata and National Research Council (CONICET), J. B. Justo 4302, 7600 Mar de/Plata, Argentina (Received 18 May 1993) The phase separation of a rubber in the course of a thermosetting polymerization is associated with heat release. Mixing is endothermic as revealed by the positive value of the interaction parameter while demixing is exothermic. Differential scanning calorimetry may be used to detect heat effects related to phase separation. Experimental results are reported for an unreactive system (castor oil/epoxy resin) and reactive systems (modifier/epoxy resin/diamine) using either castor oil or epoxy-terminated copolymers of acrylonitrile and butadiene as modifiers. (Keywords: rubber-modified epoxies; phase separation; d.s.c.) Introduction represents the residual free energy of mixing (part or all A common method to toughen epoxy resins is to of it may be considered as an enthalpic contribution). dissolve rubbers or other modifiers that become phase- The interaction parameter A, expressed in units of energy separated in the course of polymerization. The resulting per unit volume, is usually a positive value. This means two-phase structure exhibits an increase in the fracture that mixing occurs due to the prevailing influence of the resistance at the expense of lower values of the elastic combinatorial part of the equation over the interaction modulus and yield stress. term. Dissimilar molecules (positive A) are thus obliged Monitoring the phase separation process is important to mix due to an entropic driving force; their intrinsic in two respects: (i) for unreactive epoxy-rubber repulsion is manifested by an endothermic effect in the formulations, i.e. -

Certificate of Analysis
National Institute of Standards & Technology Certificate of Analysis Standard Reference Material 2285 Ignitable Liquids Test Mixture This Standard Reference Material (SRM) is intended primarily for use in the calibration of chromatographic instrumentation used for the classification of an ignitable liquid residue. SRM 2285 is a solution of 15 compounds, including even carbon number aliphatic hydrocarbons from hexane to tetracosane, toluene, p-xylene, 2-ethyltoluene, 3-ethyltoluene, and 1,2,4-trimethylbenzene in methylene chloride. A unit of SRM 2285 consists of five 2 mL ampoules, each containing approximately 1.2 mL of solution. Certified Mass Fraction Values: The certified values for the 15 constituents are given in Table 1. These values are based on results obtained from the gravimetric preparation of this solution and from the analytical results determined by using gas chromatography. A NIST certified value is a value for which NIST has the highest confidence in its accuracy in that all known or suspected sources of uncertainty have been investigated or taken into account [1]. Information Concentration Values: Information values for concentrations, expressed as percent volume fractions, are provided in Table 2 for the components. An information value is considered to be a value that will be of interest and use to the SRM user, but insufficient information is available to assess adequately the uncertainty associated with the value or only a limited number of analyses were performed [1]. Expiration of Certification: The certification of SRM 2285 is valid, within the measurement uncertainty specified, until 30 March 2023, provided the SRM is handled and stored in accordance with the instructions given in this certificate (see “Instructions for Handling, Storage, and Use”). -

Safety Data Sheet
SAFETY DATA SHEET Flammable Liquid Mixture: Docosane / Dodecane / Eicosane / Hexacosane / Hexadecane / Hexane / N-Decane / N-Heptane / N-Nonane / N-Octadecane / N- Octane / Octacosane / Tetracosane / Tetradecane Section 1. Identification GHS product identifier : Flammable Liquid Mixture: Docosane / Dodecane / Eicosane / Hexacosane / Hexadecane / Hexane / N-Decane / N-Heptane / N-Nonane / N-Octadecane / N-Octane / Octacosane / Tetracosane / Tetradecane Other means of : Not available. identification Product use : Synthetic/Analytical chemistry. SDS # : 019735 Supplier's details : Airgas USA, LLC and its affiliates 259 North Radnor-Chester Road Suite 100 Radnor, PA 19087-5283 1-610-687-5253 24-hour telephone : 1-866-734-3438 Section 2. Hazards identification OSHA/HCS status : This material is considered hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200). Classification of the : FLAMMABLE LIQUIDS - Category 1 substance or mixture SKIN CORROSION/IRRITATION - Category 2 SERIOUS EYE DAMAGE/ EYE IRRITATION - Category 2A TOXIC TO REPRODUCTION (Fertility) - Category 2 TOXIC TO REPRODUCTION (Unborn child) - Category 2 SPECIFIC TARGET ORGAN TOXICITY (SINGLE EXPOSURE) (Respiratory tract irritation) - Category 3 SPECIFIC TARGET ORGAN TOXICITY (SINGLE EXPOSURE) (Narcotic effects) - Category 3 SPECIFIC TARGET ORGAN TOXICITY (REPEATED EXPOSURE) - Category 2 AQUATIC HAZARD (ACUTE) - Category 2 AQUATIC HAZARD (LONG-TERM) - Category 1 GHS label elements Hazard pictograms : Signal word : Danger Hazard statements : Extremely flammable liquid and vapor. May form explosive mixtures in Air. Causes serious eye irritation. Causes skin irritation. May cause respiratory irritation. Suspected of damaging fertility or the unborn child. May cause drowsiness and dizziness. May cause damage to organs through prolonged or repeated exposure. Very toxic to aquatic life with long lasting effects. Precautionary statements General : Read label before use. -

Vapor Pressures and Vaporization Enthalpies of the N-Alkanes from 2 C21 to C30 at T ) 298.15 K by Correlation Gas Chromatography
BATCH: je1a04 USER: jeh69 DIV: @xyv04/data1/CLS_pj/GRP_je/JOB_i01/DIV_je0301747 DATE: October 17, 2003 1 Vapor Pressures and Vaporization Enthalpies of the n-Alkanes from 2 C21 to C30 at T ) 298.15 K by Correlation Gas Chromatography 3 James S. Chickos* and William Hanshaw 4 Department of Chemistry and Biochemistry, University of MissourisSt. Louis, St. Louis, Missouri 63121 5 6 The temperature dependence of gas chromatographic retention times for n-heptadecane to n-triacontane 7 is reported. These data are used to evaluate the vaporization enthalpies of these compounds at T ) 298.15 8 K, and a protocol is described that provides vapor pressures of these n-alkanes from T ) 298.15 to 575 9 K. The vapor pressure and vaporization enthalpy results obtained are compared with existing literature 10 data where possible and found to be internally consistent. Sublimation enthalpies for n-C17 to n-C30 are 11 calculated by combining vaporization enthalpies with fusion enthalpies and are compared when possible 12 to direct measurements. 13 14 Introduction 15 The n-alkanes serve as excellent standards for the 16 measurement of vaporization enthalpies of hydrocarbons.1,2 17 Recently, the vaporization enthalpies of the n-alkanes 18 reported in the literature were examined and experimental 19 values were selected on the basis of how well their 20 vaporization enthalpies correlated with their enthalpies of 21 transfer from solution to the gas phase as measured by gas 22 chromatography.3 A plot of the vaporization enthalpies of 23 the n-alkanes as a function of the number of carbon atoms 24 is given in Figure 1. -
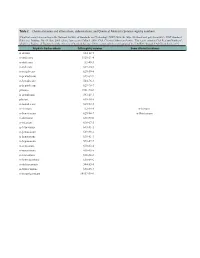
Table 2. Chemical Names and Alternatives, Abbreviations, and Chemical Abstracts Service Registry Numbers
Table 2. Chemical names and alternatives, abbreviations, and Chemical Abstracts Service registry numbers. [Final list compiled according to the National Institute of Standards and Technology (NIST) Web site (http://webbook.nist.gov/chemistry/); NIST Standard Reference Database No. 69, June 2005 release, last accessed May 9, 2008. CAS, Chemical Abstracts Service. This report contains CAS Registry Numbers®, which is a Registered Trademark of the American Chemical Society. CAS recommends the verification of the CASRNs through CAS Client ServicesSM] Aliphatic hydrocarbons CAS registry number Some alternative names n-decane 124-18-5 n-undecane 1120-21-4 n-dodecane 112-40-3 n-tridecane 629-50-5 n-tetradecane 629-59-4 n-pentadecane 629-62-9 n-hexadecane 544-76-3 n-heptadecane 629-78-7 pristane 1921-70-6 n-octadecane 593-45-3 phytane 638-36-8 n-nonadecane 629-92-5 n-eicosane 112-95-8 n-Icosane n-heneicosane 629-94-7 n-Henicosane n-docosane 629-97-0 n-tricosane 638-67-5 n-tetracosane 643-31-1 n-pentacosane 629-99-2 n-hexacosane 630-01-3 n-heptacosane 593-49-7 n-octacosane 630-02-4 n-nonacosane 630-03-5 n-triacontane 638-68-6 n-hentriacontane 630-04-6 n-dotriacontane 544-85-4 n-tritriacontane 630-05-7 n-tetratriacontane 14167-59-0 Table 2. Chemical names and alternatives, abbreviations, and Chemical Abstracts Service registry numbers.—Continued [Final list compiled according to the National Institute of Standards and Technology (NIST) Web site (http://webbook.nist.gov/chemistry/); NIST Standard Reference Database No.