The Phase Equilibrium of Alkanes and Supercritical Fluids
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Section 13. Disposal Considerations Disposal Methods : the Generation of Waste Should Be Avoided Or Minimized Wherever Possible
SAFETY DATA SHEET Flammable Liquid Mixture: Docosane / Dodecane / Dotriacontane / Eicosane / Heptane / Hexacosane / Hexadecane / Hexane / N-Decane / N-Nonane / N- Octadecane / Octacosane / Octane / Tetracosane / Tetradecane / Triacontane Section 1. Identification GHS product identifier : Flammable Liquid Mixture: Docosane / Dodecane / Dotriacontane / Eicosane / Heptane / Hexacosane / Hexadecane / Hexane / N-Decane / N-Nonane / N-Octadecane / Octacosane / Octane / Tetracosane / Tetradecane / Triacontane Other means of : Not available. identification Product use : Synthetic/Analytical chemistry. SDS # : 019981 Supplier's details : Airgas USA, LLC and its affiliates 259 North Radnor-Chester Road Suite 100 Radnor, PA 19087-5283 1-610-687-5253 24-hour telephone : 1-866-734-3438 Section 2. Hazards identification OSHA/HCS status : This material is considered hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200). Classification of the : FLAMMABLE LIQUIDS - Category 1 substance or mixture SKIN CORROSION/IRRITATION - Category 2 SERIOUS EYE DAMAGE/ EYE IRRITATION - Category 2A TOXIC TO REPRODUCTION (Fertility) - Category 2 TOXIC TO REPRODUCTION (Unborn child) - Category 2 SPECIFIC TARGET ORGAN TOXICITY (SINGLE EXPOSURE) (Narcotic effects) - Category 3 SPECIFIC TARGET ORGAN TOXICITY (REPEATED EXPOSURE) - Category 2 AQUATIC HAZARD (ACUTE) - Category 2 AQUATIC HAZARD (LONG-TERM) - Category 1 GHS label elements Hazard pictograms : Signal word : Danger Hazard statements : Extremely flammable liquid and vapor. May form explosive mixtures in Air. Causes serious eye irritation. Causes skin irritation. Suspected of damaging fertility or the unborn child. May cause drowsiness and dizziness. May cause damage to organs through prolonged or repeated exposure. Very toxic to aquatic life with long lasting effects. Precautionary statements General : Read label before use. Keep out of reach of children. If medical advice is needed, have product container or label at hand. -

Synthetic Turf Scientific Advisory Panel Meeting Materials
California Environmental Protection Agency Office of Environmental Health Hazard Assessment Synthetic Turf Study Synthetic Turf Scientific Advisory Panel Meeting May 31, 2019 MEETING MATERIALS THIS PAGE LEFT BLANK INTENTIONALLY Office of Environmental Health Hazard Assessment California Environmental Protection Agency Agenda Synthetic Turf Scientific Advisory Panel Meeting May 31, 2019, 9:30 a.m. – 4:00 p.m. 1001 I Street, CalEPA Headquarters Building, Sacramento Byron Sher Auditorium The agenda for this meeting is given below. The order of items on the agenda is provided for general reference only. The order in which items are taken up by the Panel is subject to change. 1. Welcome and Opening Remarks 2. Synthetic Turf and Playground Studies Overview 4. Synthetic Turf Field Exposure Model Exposure Equations Exposure Parameters 3. Non-Targeted Chemical Analysis Volatile Organics on Synthetic Turf Fields Non-Polar Organics Constituents in Crumb Rubber Polar Organic Constituents in Crumb Rubber 5. Public Comments: For members of the public attending in-person: Comments will be limited to three minutes per commenter. For members of the public attending via the internet: Comments may be sent via email to [email protected]. Email comments will be read aloud, up to three minutes each, by staff of OEHHA during the public comment period, as time allows. 6. Further Panel Discussion and Closing Remarks 7. Wrap Up and Adjournment Agenda Synthetic Turf Advisory Panel Meeting May 31, 2019 THIS PAGE LEFT BLANK INTENTIONALLY Office of Environmental Health Hazard Assessment California Environmental Protection Agency DRAFT for Discussion at May 2019 SAP Meeting. Table of Contents Synthetic Turf and Playground Studies Overview May 2019 Update ..... -

Sampling Dynamics for Volatile Organic Compounds Using Headspace Solid-Phase Microextraction Arrow for Microbiological Samples
separations Communication Sampling Dynamics for Volatile Organic Compounds Using Headspace Solid-Phase Microextraction Arrow for Microbiological Samples Kevin E. Eckert 1, David O. Carter 2 and Katelynn A. Perrault 1,* 1 Laboratory of Forensic and Bioanalytical Chemistry, Forensic Sciences Unit, Division of Natural Sciences and Mathematics, Chaminade University of Honolulu, 3140 Waialae Ave., Honolulu, HI 96816, USA; [email protected] 2 Laboratory of Forensic Taphonomy, Forensic Sciences Unit, Division of Natural Sciences and Mathematics, Chaminade University of Honolulu, 3140 Waialae Avenue, Honolulu, HI 96816, USA; [email protected] * Correspondence: [email protected] Received: 10 August 2018; Accepted: 27 August 2018; Published: 10 September 2018 Abstract: Volatile organic compounds (VOCs) are monitored in numerous fields using several commercially-available sampling options. Sorbent-based sampling techniques, such as solid-phase microextraction (SPME), provide pre-concentration and focusing of VOCs prior to gas chromatography–mass spectrometry (GC–MS) analysis. This study investigated the dynamics of SPME Arrow, which exhibits an increased sorbent phase volume and improved durability compared to traditional SPME fibers. A volatile reference mixture (VRM) and saturated alkanes mix (SAM) were used to investigate optimal parameters for microbiological VOC profiling in combination with GC–MS analysis. Fiber type, extraction time, desorption time, carryover, and reproducibility were characterized, in addition to a comparison with traditional SPME fibers. The developed method was then applied to longitudinal monitoring of Bacillus subtilis cultures, which represents a ubiquitous microbe in medical, forensic, and agricultural applications. The carbon wide range/polydimethylsiloxane (CWR/PDMS) fiber was found to be optimal for the range of expected VOCs in microbiological profiling, and a statistically significant increase in the majority of VOCs monitored was observed. -

Certificate of Analysis
National Institute of Standards & Technology Certificate of Analysis Standard Reference Material 2285 Ignitable Liquids Test Mixture This Standard Reference Material (SRM) is intended primarily for use in the calibration of chromatographic instrumentation used for the classification of an ignitable liquid residue. SRM 2285 is a solution of 15 compounds, including even carbon number aliphatic hydrocarbons from hexane to tetracosane, toluene, p-xylene, 2-ethyltoluene, 3-ethyltoluene, and 1,2,4-trimethylbenzene in methylene chloride. A unit of SRM 2285 consists of five 2 mL ampoules, each containing approximately 1.2 mL of solution. Certified Mass Fraction Values: The certified values for the 15 constituents are given in Table 1. These values are based on results obtained from the gravimetric preparation of this solution and from the analytical results determined by using gas chromatography. A NIST certified value is a value for which NIST has the highest confidence in its accuracy in that all known or suspected sources of uncertainty have been investigated or taken into account [1]. Information Concentration Values: Information values for concentrations, expressed as percent volume fractions, are provided in Table 2 for the components. An information value is considered to be a value that will be of interest and use to the SRM user, but insufficient information is available to assess adequately the uncertainty associated with the value or only a limited number of analyses were performed [1]. Expiration of Certification: The certification of SRM 2285 is valid, within the measurement uncertainty specified, until 30 March 2023, provided the SRM is handled and stored in accordance with the instructions given in this certificate (see “Instructions for Handling, Storage, and Use”). -

Advanced Separation Techniques Combined with Mass Spectrometry for Difficult Analytical Tasks - Isomer Separation and Oil Analysis
FINNISH METEOROLOGICAL INSTITUTE Erik Palménin aukio 1 P.O. Box 503 FI-00101 HELSINKI 131 CONTRIBUTIONS tel. +358 29 539 1000 WWW.FMI.FI FINNISH METEOROLOGICAL INSTITUTE CONTRIBUTIONS No. 131 ISBN 978-952-336-016-7 (paperback) ISSN 0782-6117 Erweko Helsinki 2017 ISBN 978-952-336-017-4 (pdf) Helsinki 2017 ADVANCED SEPARATION TECHNIQUES COMBINED WITH MASS SPECTROMETRY FOR DIFFICULT ANALYTICAL TASKS - ISOMER SEPARATION AND OIL ANALYSIS JAAKKO LAAKIA Department of Chemistry University of Helsinki Finland Advanced separation techniques combined with mass spectrometry for difficult analytical tasks – isomer separation and oil analysis Jaakko Laakia Academic Dissertation To be presented, with the permission of the Faculty of Science of the University of Helsinki, for public criticism in Auditorium A129 of the Department of Chemistry (A.I. Virtasen aukio 1, Helsinki) on May 12th, 2017, at 12 o’clock noon. Helsinki 2017 1 TABLE OF CONTENTS ABSTRACT 4 Supervisors Professor Tapio Kotiaho TIIVISTELMÄ 5 Department of Chemistry LIST OF ORIGINAL PAPERS 6 Faculty of Science ACKNOWLEDGEMENTS 7 ABBREVIATIONS 8 and 1. INTRODUCTION 10 Division of Pharmaceutical Chemistry and Technology 1.1.1 Basic operation principles of ion mobility 12 spectrometry 11 Faculty of Pharmacy 1.1.2 Applications of ion mobility spectrometry 16 University of Helsinki 1.2 Basic operation principles and applications of two-dimensional gas 19 chromatography – time-of-flight mass spectrometry Docent Tiina Kauppila 1.3 Aims of study 23 Division of Pharmaceutical Chemistry and Technology -

Safety Data Sheet
SAFETY DATA SHEET Flammable Liquid Mixture: Docosane / Dodecane / Eicosane / Hexacosane / Hexadecane / Hexane / N-Decane / N-Heptane / N-Nonane / N-Octadecane / N- Octane / Octacosane / Tetracosane / Tetradecane Section 1. Identification GHS product identifier : Flammable Liquid Mixture: Docosane / Dodecane / Eicosane / Hexacosane / Hexadecane / Hexane / N-Decane / N-Heptane / N-Nonane / N-Octadecane / N-Octane / Octacosane / Tetracosane / Tetradecane Other means of : Not available. identification Product use : Synthetic/Analytical chemistry. SDS # : 019735 Supplier's details : Airgas USA, LLC and its affiliates 259 North Radnor-Chester Road Suite 100 Radnor, PA 19087-5283 1-610-687-5253 24-hour telephone : 1-866-734-3438 Section 2. Hazards identification OSHA/HCS status : This material is considered hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200). Classification of the : FLAMMABLE LIQUIDS - Category 1 substance or mixture SKIN CORROSION/IRRITATION - Category 2 SERIOUS EYE DAMAGE/ EYE IRRITATION - Category 2A TOXIC TO REPRODUCTION (Fertility) - Category 2 TOXIC TO REPRODUCTION (Unborn child) - Category 2 SPECIFIC TARGET ORGAN TOXICITY (SINGLE EXPOSURE) (Respiratory tract irritation) - Category 3 SPECIFIC TARGET ORGAN TOXICITY (SINGLE EXPOSURE) (Narcotic effects) - Category 3 SPECIFIC TARGET ORGAN TOXICITY (REPEATED EXPOSURE) - Category 2 AQUATIC HAZARD (ACUTE) - Category 2 AQUATIC HAZARD (LONG-TERM) - Category 1 GHS label elements Hazard pictograms : Signal word : Danger Hazard statements : Extremely flammable liquid and vapor. May form explosive mixtures in Air. Causes serious eye irritation. Causes skin irritation. May cause respiratory irritation. Suspected of damaging fertility or the unborn child. May cause drowsiness and dizziness. May cause damage to organs through prolonged or repeated exposure. Very toxic to aquatic life with long lasting effects. Precautionary statements General : Read label before use. -

Vapor Pressures and Vaporization Enthalpies of the N-Alkanes from 2 C21 to C30 at T ) 298.15 K by Correlation Gas Chromatography
BATCH: je1a04 USER: jeh69 DIV: @xyv04/data1/CLS_pj/GRP_je/JOB_i01/DIV_je0301747 DATE: October 17, 2003 1 Vapor Pressures and Vaporization Enthalpies of the n-Alkanes from 2 C21 to C30 at T ) 298.15 K by Correlation Gas Chromatography 3 James S. Chickos* and William Hanshaw 4 Department of Chemistry and Biochemistry, University of MissourisSt. Louis, St. Louis, Missouri 63121 5 6 The temperature dependence of gas chromatographic retention times for n-heptadecane to n-triacontane 7 is reported. These data are used to evaluate the vaporization enthalpies of these compounds at T ) 298.15 8 K, and a protocol is described that provides vapor pressures of these n-alkanes from T ) 298.15 to 575 9 K. The vapor pressure and vaporization enthalpy results obtained are compared with existing literature 10 data where possible and found to be internally consistent. Sublimation enthalpies for n-C17 to n-C30 are 11 calculated by combining vaporization enthalpies with fusion enthalpies and are compared when possible 12 to direct measurements. 13 14 Introduction 15 The n-alkanes serve as excellent standards for the 16 measurement of vaporization enthalpies of hydrocarbons.1,2 17 Recently, the vaporization enthalpies of the n-alkanes 18 reported in the literature were examined and experimental 19 values were selected on the basis of how well their 20 vaporization enthalpies correlated with their enthalpies of 21 transfer from solution to the gas phase as measured by gas 22 chromatography.3 A plot of the vaporization enthalpies of 23 the n-alkanes as a function of the number of carbon atoms 24 is given in Figure 1. -
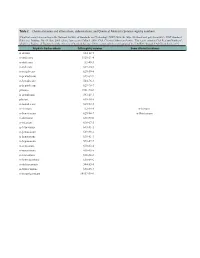
Table 2. Chemical Names and Alternatives, Abbreviations, and Chemical Abstracts Service Registry Numbers
Table 2. Chemical names and alternatives, abbreviations, and Chemical Abstracts Service registry numbers. [Final list compiled according to the National Institute of Standards and Technology (NIST) Web site (http://webbook.nist.gov/chemistry/); NIST Standard Reference Database No. 69, June 2005 release, last accessed May 9, 2008. CAS, Chemical Abstracts Service. This report contains CAS Registry Numbers®, which is a Registered Trademark of the American Chemical Society. CAS recommends the verification of the CASRNs through CAS Client ServicesSM] Aliphatic hydrocarbons CAS registry number Some alternative names n-decane 124-18-5 n-undecane 1120-21-4 n-dodecane 112-40-3 n-tridecane 629-50-5 n-tetradecane 629-59-4 n-pentadecane 629-62-9 n-hexadecane 544-76-3 n-heptadecane 629-78-7 pristane 1921-70-6 n-octadecane 593-45-3 phytane 638-36-8 n-nonadecane 629-92-5 n-eicosane 112-95-8 n-Icosane n-heneicosane 629-94-7 n-Henicosane n-docosane 629-97-0 n-tricosane 638-67-5 n-tetracosane 643-31-1 n-pentacosane 629-99-2 n-hexacosane 630-01-3 n-heptacosane 593-49-7 n-octacosane 630-02-4 n-nonacosane 630-03-5 n-triacontane 638-68-6 n-hentriacontane 630-04-6 n-dotriacontane 544-85-4 n-tritriacontane 630-05-7 n-tetratriacontane 14167-59-0 Table 2. Chemical names and alternatives, abbreviations, and Chemical Abstracts Service registry numbers.—Continued [Final list compiled according to the National Institute of Standards and Technology (NIST) Web site (http://webbook.nist.gov/chemistry/); NIST Standard Reference Database No. -

Exhaust Emission Profiles for EPA SPECIATE Database
Exhaust Emission Profiles for EPA SPECIATE Database: Energy Policy Act (EPAct) Low-Level Ethanol Fuel Blends and Tier 2 Light- Duty Vehicles Exhaust Emission Profiles for EPA SPECIATE Database: Energy Policy Act (EPAct) Low-Level Ethanol Fuel Blends and Tier 2 Light- Duty Vehicles Assessment and Standards Division Office of ransportationT and Air Quality U.S. Environmental Protection Agency EPA-420-R-09-002 June 2009 Table of Contents 1.0 Introduction....................................................................................................................... 1 1.1 Background..................................................................................................................... 1 1.2 Purpose of the Project ..................................................................................................... 1 2.0 Methods.............................................................................................................................. 2 2.1 Exhaust Emissions Data.................................................................................................. 2 2.2 Calculation of Composite Profiles.................................................................................. 3 2.3 Adjustment of E0 and E10 profiles with an In-Use Gasoline......................................... 3 2.4 Assignment of SPECIATE Identification Numbers ....................................................... 4 3.0 Results ............................................................................................................................... -

Discovery of Optimal Zeolites for Challenging Separations and Chemical Transformations Using Predictive Materials Modeling
ARTICLE Received 9 Jul 2014 | Accepted 19 Nov 2014 | Published 21 Jan 2015 DOI: 10.1038/ncomms6912 Discovery of optimal zeolites for challenging separations and chemical transformations using predictive materials modeling Peng Bai1, Mi Young Jeon1, Limin Ren1, Chris Knight2, Michael W. Deem3, Michael Tsapatsis1 & J. Ilja Siepmann1 Zeolites play numerous important roles in modern petroleum refineries and have the potential to advance the production of fuels and chemical feedstocks from renewable resources. The performance of a zeolite as separation medium and catalyst depends on its framework structure. To date, 213 framework types have been synthesized and 4330,000 thermo- dynamically accessible zeolite structures have been predicted. Hence, identification of optimal zeolites for a given application from the large pool of candidate structures is attractive for accelerating the pace of materials discovery. Here we identify, through a large- scale, multi-step computational screening process, promising zeolite structures for two energy-related applications: the purification of ethanol from fermentation broths and the hydroisomerization of alkanes with 18–30 carbon atoms encountered in petroleum refining. These results demonstrate that predictive modelling and data-driven science can now be applied to solve some of the most challenging separation problems involving highly non-ideal mixtures and highly articulated compounds. 1 Departments of Chemistry and of Chemical Engineering and Materials Science and Chemical Theory Center, University of Minnesota, 207 Pleasant Street S.E., Minneapolis, Minnesota 55455, USA. 2 Leadership Computing Facility, Argonne National Laboratory, 9700 S. Cass Avenue, Argonne, Illinois 60439, USA. 3 Departments of Bioengineering and of Physics and Astronomy, Rice University, 6100 Main Street, Houston, Texas 77005, USA. -

Hydrogenated Poly-1-Decene
HYDROGENATED POLY-1-DECENE Prepared at the 57th JECFA (2001) and published in FNP 52 Add 9 (2001), superseding specifications prepared at the 53rd JECFA (1999), published in FNP 52 Add 7 (1999). An ADI of 0 - 6 mg/kg bw was established at the 57th JECFA (2001). SYNONYMS Hydrogenated polydec-1-ene, Hydrogenated poly-α-olefin; INS No. 907 DEFINITION Hydrogenated poly-1-decene is a mixture of branched isomeric hydrocarbons, prepared by hydrogenation of mixtures of trimers, tetramers, pentamers and hexamers of 1-decenes. Minor amounts of molecules with carbon number less than 30 may be present. C.A.S. number 68037-01-4 Chemical formula C10nH20n+2, where n = 3 – 6 Formula weight 560 (average) Assay Not less than 98.5% of hydrogenated poly-1-decene, having the following oligomer distribution: 3 C 0: 13 - 37% C40: 35 - 70% C50: 9 - 25% C60: 1 - 7% DESCRIPTION Colourless, odourless, viscous liquid FUNCTIONAL USES Glazing agent, release agent CHARACTERISTICS IDENTIFICATION Solubility (Vol. 4) Insoluble in water; slightly soluble in ethanol; soluble in toluene Burning The product burns with bright flame and a paraffin-like characteristic smell Viscosity 5.7 - 6.1 mm2/sec (100°) See description under TESTS PURITY Compounds with carbon Not more than 1.5 % number less than 30 See METHOD OF ASSAY Readily carbonizable Passes test substances See description under TESTS Nickel (Vol. 4) Not more than 1 mg/kg Lead (Vol. 4) Not more than 1 mg/kg Determine using an atomic absorption technique appropriate to the specified level. The selection of sample size and method of sample preparation may be based on the principles of the method described on Volume 4, "Instrumental methods". -

ABSTRACT RUDD, HAYDEN. Assessing the Vulnerability Of
ABSTRACT RUDD, HAYDEN. Assessing the Vulnerability of Coastal Plain Groundwater to Flood Water Intrusion using High Resolution Mass Spectrometry. (Under the direction of Dr. Elizabeth Guthrie Nichols). Communities in the North Carolina Coastal Plain (NCCP) depend on safe and reliable groundwater for private well use, agriculture, industry, and livelihoods. Although storm intensity and frequency are predicted to increase in coastal areas, the risk of surficial and confined aquifer contamination from extreme storms is not understood. In September 2018, Hurricane Florence caused extensive flooding across the NCCP for several weeks. The North Carolina Department of Environmental Quality (NCDEQ) Groundwater Management Branch had just completed sampling of some wells in their monitoring network when Hurricane Florence made landfall. NCDEQ returned to these wells, particularly those flooded by Hurricane Florence, for post-flood sampling. These groundwater samples were analyzed by NCDEQ for regulated semi-volatile organics with few to any detections of regulated organic contaminants. NCDEQ provided NC State the same sample extracts for analysis by high resolution mass spectrometry (HRMS). This research reports on the non-targeted and suspect-screening HRMS analyses of groundwater from nested monitoring wells. Some monitoring well sites experienced flooding during the study period, and some did not. The goal of this research was to advance our understanding of coastal aquifer susceptibility to flooding by producing the first comprehensive organic chemical profiles of coastal aquifers and by determining if aquifers have distinct organic chemical profiles that change after flooding events. This study used HRMS analyses to produce the first comprehensive organic chemical profiles of 11 aquifers in the coastal plain.