Contents • Introduction to Proteomics and Mass Spectrometry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mass Spectrometer Business Presentation Materials
Mass Spectrometer Business Presentation Materials Hiroto Itoi, Corporate Officer Deputy General Manager of the Analytical & Measuring Instruments Division Shimadzu Corporation Jul. 3, 2018 Contents I. Introduction • Expansion of Mass Spectrometry ………………………………………………………………… p.3 • History of Shimadzu's Growth in Mass Spectrometry …………………………………………… p.5 II. Overview of Mass Spectrometers • Operating Principle, Demand Trends, and Vendors ……………………………………………… p.9 • Mass Spectra ………………………………………………………………………………………… p.10 • Configuration of Mass Spectrometers …………………………………………………………… p.11 • Ionization …………………………………………………………………………………………… p.12 • Mass Separation …………………………………………………………………………………… p.14 III. Shimadzu's Mass Spectrometer Business • Product Type ………………………………………………………………………………………… p.17 • Application Software ………………………………………………………………………………… p.18 • Growth Strategy for Mass Spectrometer Business ……………………………………………… p.19 • Expand/Improve Product Lines …………………………………………………………………… p.20 • Measures to Expand Application Fields …………………………………………………………… p.24 • Measures to Automate Data Processing Using AI ……………………………………………… p.25 IV. Summary • Future Direction ……………………………………………………………………………………… p.26 July 2018 Mass Spectrometer Business Presentation Materials 2 I. Introduction Expansion of Mass Spectrometry (1) Why Mass Spectrometry? Mass spectrometry is able to analyze a wide variety of compounds with high accuracy and high efficiency (simultaneous multicomponent analysis). It offers superior characteristics that are especially beneficial in the following fields, -

(ESI) Electrospray Ionization
Electrospray Ionization (ESI) • sample solution • voltage drop • solvent evaporates is injected between source, away as droplets directly into analyzer generates migrate across gap instrument ionized droplets “sheath gas” Electrospray Ionization (ESI) Injected solution usually includes ionizing agent H+ (from acid), Na+, K+ Large droplets from injected stream Resulting ions can be spontaneously eject singly or multiply smaller droplets (due to charged electrostatic repulsion) Electrospray Ionization (ESI) Multiply charged ions frequently observed. ESI M [M•H]+ + [M•2H]2+ + [M•3H]3+ + ... or [M•Na]+ + [M•2Na]2+ + [M•3Na]3+ + ... multiply lysozyme: charged big > 10 kDa protein (protonated) ions From this series of peaks, how do we determine mass of protein? Charge Ladders in Electrospray Ionization We know two things: 1. Any peak corresponds to [M•nH]n+; ion mass m =M+= M + n, charge z = n. 2. Peak to that peak’s left has one more proton attached, one more charge; ion mass m = M + n + 1, charge z = n + 1. Charge Ladders in Electrospray Ionization Set of simultaneous equations: (observed m/z #1) • z = M + z(H+) (observed m/z #2) • (z + 1) = M + z(H+) + 1 2 1 Charge Ladders in Electrospray Ionization Now performed by computer algorithm Set of simultaneous equations: that searches for ladders, outputs likely mass candidates. 1486 • z = M + z 1372.5 • (z + 1) = M + z + 1 Solution: z = 12 M = 17827.2 [M•13H]13+ 2 [M•12H]1 12+ actual MW lysozyme: 17825.2 If sample contains a mixture of counterions, can get messy. ESI-MS 22-mer single-stranded DNA (difficult to desalt) Atmospheric Pressure Chemical Ionization (APCI) •+ •+ Analogous to CI, but uses discharge to generate N2 /O2 , which then iiionizes evapora tdltllThthiited solvent molecules. -

Modern Mass Spectrometry
Modern Mass Spectrometry MacMillan Group Meeting 2005 Sandra Lee Key References: E. Uggerud, S. Petrie, D. K. Bohme, F. Turecek, D. Schröder, H. Schwarz, D. Plattner, T. Wyttenbach, M. T. Bowers, P. B. Armentrout, S. A. Truger, T. Junker, G. Suizdak, Mark Brönstrup. Topics in Current Chemistry: Modern Mass Spectroscopy, pp. 1-302, 225. Springer-Verlag, Berlin, 2003. Current Topics in Organic Chemistry 2003, 15, 1503-1624 1 The Basics of Mass Spectroscopy ! Purpose Mass spectrometers use the difference in mass-to-charge ratio (m/z) of ionized atoms or molecules to separate them. Therefore, mass spectroscopy allows quantitation of atoms or molecules and provides structural information by the identification of distinctive fragmentation patterns. The general operation of a mass spectrometer is: "1. " create gas-phase ions "2. " separate the ions in space or time based on their mass-to-charge ratio "3. " measure the quantity of ions of each mass-to-charge ratio Ionization sources ! Instrumentation Chemical Ionisation (CI) Atmospheric Pressure CI!(APCI) Electron Impact!(EI) Electrospray Ionization!(ESI) SORTING DETECTION IONIZATION OF IONS OF IONS Fast Atom Bombardment (FAB) Field Desorption/Field Ionisation (FD/FI) Matrix Assisted Laser Desorption gaseous mass ion Ionisation!(MALDI) ion source analyzer transducer Thermospray Ionisation (TI) Analyzers quadrupoles vacuum signal Time-of-Flight (TOF) pump processor magnetic sectors 10-5– 10-8 torr Fourier transform and quadrupole ion traps inlet Detectors mass electron multiplier spectrum Faraday cup Ionization Sources: Classical Methods ! Electron Impact Ionization A beam of electrons passes through a gas-phase sample and collides with neutral analyte molcules (M) to produce a positively charged ion or a fragment ion. -
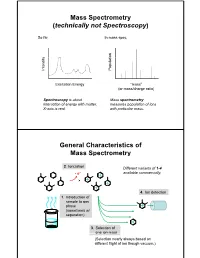
Mass Spectrometry (Technically Not Spectroscopy)
Mass Spectrometry (technically not Spectroscopy) So far, In mass spec, on y Populati Intensit Excitation Energy “mass” (or mass/charge ratio) Spectroscopy is about Mass spectrometry interaction of energy with matter. measures population of ions X-axis is real. with particular mass. General Characteristics of Mass Spectrometry 2. Ionization Different variants of 1-4 -e- available commercially. 4. Ion detection 1. Introduction of sample to gas phase (sometimes w/ separation) 3. Selection of one ion mass (Selection nearly always based on different flight of ion though vacuum.) General Components of a Mass Spectrometer Lots of choices, which can be mixed and matched. direct injection The Mass Spectrum fragment “daughter” ions M+ “parent” mass Sample Introduction: Direc t Inser tion Prob e If sample is a liquid, sample can also be injected directly into ionization region. If sample isn’t pure, get multiple parents (that can’t be distinguished from fragments). Capillary Column Introduction Continous source of molecules to spectrometer. detector column (including GC, LC, chiral, size exclusion) • Signal intensity depends on both amount of molecule and ionization efficiency • To use quantitatively, must calibrate peaks with respect eltilution time ttlitotal ion curren t to quantity eluted (TIC) over time Capillary Column Introduction Easy to interface with gas or liquid chromatography. TIC trace elution time time averaged time averaged mass spectrum mass spectrum Methods of Ionization: Electron Ionization (EI) 1 - + - 1 M + e (kV energy) M + 2e Fragmentation in Electron Ionization daughter ion (observed in spectrum) neutral fragment (not observed) excited parent at electron at electron energy of energy of 15 eV 70 e V Lower electron energy yields less fragmentation, but also less signal. -

An Introduction to Mass Spectrometry
An Introduction to Mass Spectrometry by Scott E. Van Bramer Widener University Department of Chemistry One University Place Chester, PA 19013 [email protected] http://science.widener.edu/~svanbram revised: September 2, 1998 © Copyright 1997 TABLE OF CONTENTS INTRODUCTION ........................................................... 4 SAMPLE INTRODUCTION ....................................................5 Direct Vapor Inlet .......................................................5 Gas Chromatography.....................................................5 Liquid Chromatography...................................................6 Direct Insertion Probe ....................................................6 Direct Ionization of Sample ................................................6 IONIZATION TECHNIQUES...................................................6 Electron Ionization .......................................................7 Chemical Ionization ..................................................... 9 Fast Atom Bombardment and Secondary Ion Mass Spectrometry .................10 Atmospheric Pressure Ionization and Electrospray Ionization ....................11 Matrix Assisted Laser Desorption/Ionization ................................ 13 Other Ionization Methods ................................................13 Self-Test #1 ...........................................................14 MASS ANALYZERS .........................................................14 Quadrupole ............................................................15 -

An Organic Chemist's Guide to Electrospray Mass Spectrometric
molecules Review An Organic Chemist’s Guide to Electrospray Mass Spectrometric Structure Elucidation Arnold Steckel 1 and Gitta Schlosser 2,* 1 Hevesy György PhD School of Chemistry, ELTE Eötvös Loránd University, Pázmány Péter sétány 1/A, 1117 Budapest, Hungary; [email protected] 2 Department of Analytical Chemistry, ELTE Eötvös Loránd University, Pázmány Péter sétány 1/A, 1117 Budapest, Hungary * Correspondence: [email protected] Received: 16 January 2019; Accepted: 8 February 2019; Published: 10 February 2019 Abstract: Tandem mass spectrometry is an important tool for structure elucidation of natural and synthetic organic products. Fragmentation of odd electron ions (OE+) generated by electron ionization (EI) was extensively studied in the last few decades, however there are only a few systematic reviews available concerning the fragmentation of even-electron ions (EE+/EE−) produced by the currently most common ionization techniques, electrospray ionization (ESI) and atmospheric pressure chemical ionization (APCI). This review summarizes the most important features of tandem mass spectra generated by collision-induced dissociation fragmentation and presents didactic examples for the unexperienced users. Keywords: tandem mass spectrometry; MS/MS fragmentation; collision-induced dissociation; CID; ESI; structure elucidation 1. Introduction Electron ionization (EI), a hard ionization technique, is the method of choice for analyses of small (<1000 Da), nonpolar, volatile compounds. As its name implies, the technique involves ionization by electrons with ~70 eV energy. This energy is high enough to yield very reproducible mass spectra with a large number of fragments. However, these spectra frequently lack the radical type molecular ions (M+) due to the high internal energy transferred to the precursors [1]. -

Orbitrap Fusion Tribrid Mass Spectrometer
MASS SPECTROMETRY Product Specifications Thermo Scientific Orbitrap Fusion Tribrid Mass Spectrometer Unmatched analytical performance, revolutionary MS architecture The Thermo Scientific™ Orbitrap Fusion™ mass spectrometer combines the best of quadrupole, Orbitrap, and linear ion trap mass analysis in a revolutionary Thermo Scientific™ Tribrid™ architecture that delivers unprecedented depth of analysis. It enables life scientists working with even the most challenging samples—samples of low abundance, high complexity, or difficult-to-analyze chemical structure—to identify more compounds faster, quantify them more accurately, and elucidate molecular composition more thoroughly. • Tribrid architecture combines quadrupole, followed by ETD or EThCD for glycopeptide linear ion trap, and Orbitrap mass analyzers characterization or HCD followed by CID • Multiple fragmentation techniques—CID, for small-molecule structural analysis. HCD, and optional ETD and EThCD—are available at any stage of MSn, with The ultrahigh resolution of the Orbitrap mass subsequent mass analysis in either the ion analyzer increases certainty of analytical trap or Orbitrap mass analyzer results, enabling molecular-weight • Parallelization of MS and MSn acquisition determination for intact proteins and confident to maximize the amount of high-quality resolution of isobaric species. The unsurpassed data acquired scan rate and resolution of the system are • Next-generation ion sources and ion especially useful when dealing with complex optics increase system ease of operation and robustness and low-abundance samples in proteomics, • Innovative instrument control software metabolomics, glycomics, lipidomics, and makes setup easier, methods more similar applications. powerful, and operation more intuitive The intuitive user interface of the tune editor The Orbitrap Fusion Tribrid MS can perform and method editor makes instrument calibration a wide variety of analyses, from in-depth and method development easier. -

Methods of Ion Generation
Chem. Rev. 2001, 101, 361−375 361 Methods of Ion Generation Marvin L. Vestal PE Biosystems, Framingham, Massachusetts 01701 Received May 24, 2000 Contents I. Introduction 361 II. Atomic Ions 362 A. Thermal Ionization 362 B. Spark Source 362 C. Plasma Sources 362 D. Glow Discharge 362 E. Inductively Coupled Plasma (ICP) 363 III. Molecular Ions from Volatile Samples. 364 A. Electron Ionization (EI) 364 B. Chemical Ionization (CI) 365 C. Photoionization (PI) 367 D. Field Ionization (FI) 367 IV. Molecular Ions from Nonvolatile Samples 367 Marvin L. Vestal received his B.S. and M.S. degrees, 1958 and 1960, A. Spray Techniques 367 respectively, in Engineering Sciences from Purdue Univesity, Layfayette, IN. In 1975 he received his Ph.D. degree in Chemical Physics from the B. Electrospray 367 University of Utah, Salt Lake City. From 1958 to 1960 he was a Scientist C. Desorption from Surfaces 369 at Johnston Laboratories, Inc., in Layfayette, IN. From 1960 to 1967 he D. High-Energy Particle Impact 369 became Senior Scientist at Johnston Laboratories, Inc., in Baltimore, MD. E. Low-Energy Particle Impact 370 From 1960 to 1962 he was a Graduate Student in the Department of Physics at John Hopkins University. From 1967 to 1970 he was Vice F. Low-Energy Impact with Liquid Surfaces 371 President at Scientific Research Instruments, Corp. in Baltimore, MD. From G. Flow FAB 371 1970 to 1975 he was a Graduate Student and Research Instructor at the H. Laser Ionization−MALDI 371 University of Utah, Salt Lake City. From 1976 to 1981 he became I. -

A Novel High Resolution Accurate Mass Orbitrap-Based GC-MS
ROUTINE OR RESEARCH GC-Orbitrap for Environmental Analysis a collaboration between ROUTINE OR RESEARCH GC-Orbitrap for Environmental Analysis Foreword A Novel High Resolution Accurate Mass Orbitrap-based GC-MS Platform for Routine Analysis of Short Chained Chlorinated Paraffins In this study, the performance of a novel bench top, high resolution accurate mass Orbitrap™-based GC-MS was tested for the analysis of SCCPs. System performance was tested using full-scan acquisition and simple instrumental setup. Pyrolysis-GC-Orbitrap MS - A Powerful Analytical Tool for Identification and Quantification of Microplastics in a Biological Matrix The purpose of the experiments described in this work was to assess the applicability of pyrolysis-gas chromatography-Orbitrap™ mass spectrometry for the qualitative and quantitative analysis of plastic polymers in complex biological matrices. Low Level Quantification of NDMA and Non-targeted Contaminants Screening in Drinking Water using GC Orbitrap Mass Spectrometry In this work, a sensitive and selective method for NDMA detection and quantification using high resolution accurate mass GC Orbitrap™ technology is described. Overcoming Analytical Challenges for Polybrominated Diphenyl Ethers (PBDEs) Analysis in Environmental Samples using Gas Chromatography – Orbitrap Mass Spectrometry The note demonstrates the quantitative performance of the Thermo Scientific™ Exactive™ GC Orbitrap™ GC-MS mass spectrometer for the analysis of polybrominated diphenyl ethers (PBDEs) in environmental samples. Versatility of GC-Orbitrap Mass Spectrometry for the Ultra-trace Detection of Persistent Organic Pollutants in Penguin Blood from Antarctica In this study, the performance of the Thermo Scientific™ Q Exactive™ GC Orbitrap™ mass spectrometer was evaluated for routine analysis of POPs within King penguin blood from Antarctica. -

Selective Chemical Ionization of Nitrogen and Sulfur Heterocycles in Petroleum Fractions by Ion Trap Mass Spectrometry
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Selective Chemical Ionization of Nitrogen and Sulfur Heterocycles in Petroleum Fractions by Ion Trap Mass Spectrometry C. S. Creaser*, F. Krokos, and K. E. O’Neill Shod of Chemical Sciences, University of East Anglia, Norwich, United Kingdom M. J. C. Smith and P. G. McDowell BP Research, Sunbury-on-Thames, Middlesex, United Kingdom A procedure is reported for the selective ammonia chemical ionization of some nitrogen and sulfur heterocycles in petroleum fractions using ion trap mass spectrometry (ITMS). The ion trap scan routine is designed to optimize the population of ammonium reagent ions and eject from the trap (by radio frequency/direct current isolation) electron ionization products formed during reagent ion formation prior to ionization of the sample. The ITMS procedure is compared with standard ion trap detector and conventional quadrupole ammonia chemi- cal ionization for the determination of nitrogen and sulfur heterocycles in gas oil and kerosine samples. Greatly enhanced selectivity is shown for the ITMS procedure by sup res- sion of competing charge-exchange processes. (1 Am Sot Mass Spectrum 1993, 4, 322-326 ‘; he removal of organic compounds containing reagent ion NH;, is formed via the reactions [5] nitrogen and sulfur is an important part of the T petroleum-refining process 111. Their presence in NH, ‘1 NH;’ (1) petroleum distillates results in the formation of envi- NH, f NH:‘+ NH; + NH; (2) ronmental pollutants (SO,, NO,) during combustion, and their reactivity can lead to catalyst poisoning Whereas NH: is unreactive toward a wide range of in refinery processes. -

Electrification Ionization: Fundamentals and Applications
Louisiana State University LSU Digital Commons LSU Doctoral Dissertations Graduate School 11-12-2019 Electrification Ionization: undamentalsF and Applications Bijay Kumar Banstola Louisiana State University and Agricultural and Mechanical College Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_dissertations Part of the Analytical Chemistry Commons Recommended Citation Banstola, Bijay Kumar, "Electrification Ionization: undamentalsF and Applications" (2019). LSU Doctoral Dissertations. 5103. https://digitalcommons.lsu.edu/gradschool_dissertations/5103 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Doctoral Dissertations by an authorized graduate school editor of LSU Digital Commons. For more information, please [email protected]. ELECTRIFICATION IONIZATION: FUNDAMENTALS AND APPLICATIONS A Dissertation Submitted to the Graduate Faculty of the Louisiana State University and Agricultural and Mechanical College in partial fulfillment of the requirements for the degree of Doctor of Philosophy in The Department of Chemistry by Bijay Kumar Banstola B. Sc., Northwestern State University of Louisiana, 2011 December 2019 This dissertation is dedicated to my parents: Tikaram and Shova Banstola my wife: Laxmi Kandel ii ACKNOWLEDGEMENTS I thank my advisor Professor Kermit K. Murray, for his continuous support and guidance throughout my Ph.D. program. Without his unwavering guidance and continuous help and encouragement, I could not have completed this program. I am also thankful to my committee members, Professor Isiah M. Warner, Professor Kenneth Lopata, and Professor Shengli Chen. I thank Dr. Fabrizio Donnarumma for his assistance and valuable insights to overcome the hurdles throughout my program. I appreciate Miss Connie David and Dr. -

Electrospray Ionization and Samples Complexity in Meta-Metabolomics
Electrospray Ionization and samples complexity in Meta-metabolomics: a biomarker or a suppressed ion? Hikmat Ghosson, Chandrashekhar Patil, Amani Ben Jrad, Delphine Raviglione, Marie-Virginie Salvia, Cédric Bertrand To cite this version: Hikmat Ghosson, Chandrashekhar Patil, Amani Ben Jrad, Delphine Raviglione, Marie-Virginie Salvia, et al.. Electrospray Ionization and samples complexity in Meta-metabolomics: a biomarker or a suppressed ion?. European RFMF Metabomeeting 2020, Jan 2020, Toulouse, France. European RFMF Metabomeeting 2020 Book Of Abstract, pp.248, 2020, Poster abstracts. hal-02456148 HAL Id: hal-02456148 https://hal.archives-ouvertes.fr/hal-02456148 Submitted on 27 Jan 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution - NonCommercial - NoDerivatives| 4.0 International License Project co-financed by the European Regional Development Fund (ERDF) Electrospray Ionization and samples complexity in Meta-metabolomics: a biomarker or a suppressed ion? Hikmat Ghosson1,2,*, Chandrashekhar Patil1, Amani Ben Jrad1,2, Delphine Raviglione1,2, Marie-Virginie Salvia1,2,3, Cédric Bertrand1,2,3,4 *: [email protected] 1: PSL Université Paris: EPHE-UPVD-CNRS, USR 3278 CRIOBE, Université de Perpignan, 52 Avenue Paul Alduy, 66860 Perpignan Cedex, France 2: UFR Sciences Exactes et Expérimentales, Université de Perpignan Via Domitia, 52 Avenue Paul Alduy, 66860 Perpignan Cedex, France 3: Laboratoire d’Excellence « CORAIL », Université de Perpignan, 52 Avenue Paul Alduy, 66860 Perpignan Cedex, France 4: S.A.S.