Gas-Phase Ion Chemistry: Kinetics and Thermodynamics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chemistry Grade Level 10 Units 1-15
COPPELL ISD SUBJECT YEAR AT A GLANCE GRADE HEMISTRY UNITS C LEVEL 1-15 10 Program Transfer Goals ● Ask questions, recognize and define problems, and propose solutions. ● Safely and ethically collect, analyze, and evaluate appropriate data. ● Utilize, create, and analyze models to understand the world. ● Make valid claims and informed decisions based on scientific evidence. ● Effectively communicate scientific reasoning to a target audience. PACING 1st 9 Weeks 2nd 9 Weeks 3rd 9 Weeks 4th 9 Weeks Unit 1 Unit 2 Unit 3 Unit 4 Unit 5 Unit 6 Unit Unit Unit Unit Unit Unit Unit Unit Unit 7 8 9 10 11 12 13 14 15 1.5 wks 2 wks 1.5 wks 2 wks 3 wks 5.5 wks 1.5 2 2.5 2 wks 2 2 2 wks 1.5 1.5 wks wks wks wks wks wks wks Assurances for a Guaranteed and Viable Curriculum Adherence to this scope and sequence affords every member of the learning community clarity on the knowledge and skills on which each learner should demonstrate proficiency. In order to deliver a guaranteed and viable curriculum, our team commits to and ensures the following understandings: Shared Accountability: Responding -

Prebiological Evolution and the Metabolic Origins of Life
Prebiological Evolution and the Andrew J. Pratt* Metabolic Origins of Life University of Canterbury Keywords Abiogenesis, origin of life, metabolism, hydrothermal, iron Abstract The chemoton model of cells posits three subsystems: metabolism, compartmentalization, and information. A specific model for the prebiological evolution of a reproducing system with rudimentary versions of these three interdependent subsystems is presented. This is based on the initial emergence and reproduction of autocatalytic networks in hydrothermal microcompartments containing iron sulfide. The driving force for life was catalysis of the dissipation of the intrinsic redox gradient of the planet. The codependence of life on iron and phosphate provides chemical constraints on the ordering of prebiological evolution. The initial protometabolism was based on positive feedback loops associated with in situ carbon fixation in which the initial protometabolites modified the catalytic capacity and mobility of metal-based catalysts, especially iron-sulfur centers. A number of selection mechanisms, including catalytic efficiency and specificity, hydrolytic stability, and selective solubilization, are proposed as key determinants for autocatalytic reproduction exploited in protometabolic evolution. This evolutionary process led from autocatalytic networks within preexisting compartments to discrete, reproducing, mobile vesicular protocells with the capacity to use soluble sugar phosphates and hence the opportunity to develop nucleic acids. Fidelity of information transfer in the reproduction of these increasingly complex autocatalytic networks is a key selection pressure in prebiological evolution that eventually leads to the selection of nucleic acids as a digital information subsystem and hence the emergence of fully functional chemotons capable of Darwinian evolution. 1 Introduction: Chemoton Subsystems and Evolutionary Pathways Living cells are autocatalytic entities that harness redox energy via the selective catalysis of biochemical transformations. -

4.2 Ionic Bonds Vocabulary: Ion – Polyatomic Ion – Ionic Bond – Ionic Compound – Chemical Formula – Subscript –
4.2 Ionic Bonds Vocabulary: Ion – Polyatomic ion – Ionic bond – Ionic compound – Chemical formula – Subscript – Crystal - An ion is an atom or group of atoms that has an electric charge. When a neutral atom loses a valence electron, it loses a negative charge. It becomes a positive ion. When a neutral atom gains an electron, it gains a negative charge and becomes a negative ion. Common Ions: Name Charge Symbol/Formula Lithium 1+ Li+ Sodium 1+ Na+ Potassium 1+ K+ Ammonium 1+ NH₄+ Calcium 2+ Ca²+ Magnesium 2+ Mg²+ Aluminum 3+ Al³+ Fluoride 1- F- Chloride 1- Cl- Iodide 1- I- Bicarbonate 1- HCO₃- Nitrate 1- NO₃- Oxide 2- O²- Sulfide 2- S²- Carbonate 2- CO₃²- Sulfate 2- SO₄²- Notice that some ions are made of several atoms. Ammonium is made of 1 nitrogen atom and 4 hydrogen atoms. Ions that are made of more than 1 atom are called polyatomic ions. Ionic bonds: When atoms that easily lose electrons react with atoms that easily gain electrons, valence electrons are transferred from one type to another. The transfer gives each type of atom a more stable arrangement of electrons. 1. Sodium has 1 valence electron. Chlorine has 7 valence electrons. 2. The valence electron of sodium is transferred to the chlorine atom. Both atoms become ions. Sodium atom becomes a positive ion (Na+) and chlorine becomes a negative ion (Cl-). 3. Oppositely charged particles attract, so the ions attract. An ionic bond is the attraction between 2 oppositely charged ions. The resulting compound is called an ionic compound. In an ionic compound, the total overall charge is zero because the total positive charges are equal to the total negative charges. -

Analysis of Multipolar Linear Paul Traps for Ion–Atom Ultracold Collision Experiments
atoms Article Analysis of Multipolar Linear Paul Traps for Ion–Atom Ultracold Collision Experiments M. Niranjan *, Anand Prakash and S. A. Rangwala * Raman Research Institute, C. V. Raman Avenue, Sadashivanagar, Bangalore 560080, India; [email protected] * Correspondence: [email protected] (M.N.); [email protected] (S.A.R.) Abstract: We evaluate the performance of multipole, linear Paul traps for the purpose of studying cold ion–atom collisions. A combination of numerical simulations and analysis based on the virial theorem is used to draw conclusions on the differences that result, by considering the trapping details of several multipole trap types. Starting with an analysis of how a low energy collision takes place between a fully compensated, ultracold trapped ion and an stationary atom, we show that a higher order multipole trap is, in principle, advantageous in terms of collisional heating. The virial analysis of multipole traps then follows, along with the computation of trapped ion trajectories in the quadrupole, hexapole, octopole and do-decapole radio frequency traps. A detailed analysis of the motion of trapped ions as a function of the amplitude, phase and stability of the ion’s motion is used to evaluate the experimental prospects for such traps. The present analysis has the virtue of providing definitive answers for the merits of the various configurations, using first principles. Keywords: ion trapping; ion–atom collisions; linear multipole traps; virial theorem Citation: Niranjan, M.; Prakash, A.; Rangwala, S.A. Analysis of Multipolar Linear Paul Traps for 1. Introduction Ion–Atom Ultracold Collision Linear multipole Paul trap configurations are emerging as a natural choice for a wide Experiments. -

Contents • Introduction to Proteomics and Mass Spectrometry
Contents • Introduction to Proteomics and Mass spectrometry - What we need to know in our quest to explain life - Fundamental things you need to know about mass spectrometry • Interfaces and ion sources - Electrospray ionization (ESI) - conventional and nanospray - Heated nebulizer atmospheric pressure chemical ionization - Matrix assisted laser desorption • Types of MS analyzers - Magnetic sector - Quadrupole - Time-of-flight - Hybrid - Ion trap In the next step in our quest to explain what is life The human genome project has largely been completed and many other genomes are surrendering to the gene sequencers. However, all this knowledge does not give us the information that is needed to explain how living cells work. To do that, we need to study proteins. In 2002, mass spectrometry has developed to the point where it has the capacity to obtain the "exact" molecular weight of many macromolecules. At the present time, this includes proteins up to 150,000 Da. Proteins of higher molecular weights (up to 500,000 Da) can also be studied by mass spectrometry, but with less accuracy. The paradigm for sequencing of peptides and identification of proteins has changed – because of the availability of the human genome database, peptides can be identified merely by their masses or by partial sequence information, often in minutes, not hours. This new capacity is shifting the emphasis of biomedical research back to the functional aspects of cell biochemistry, the expression of particular sets of genes and their gene products, the proteins of the cell. These are the new goals of the biological scientist: o to know which proteins are expressed in each cell, preferably one cell at a time o to know how these proteins are modified, information that cannot necessarily be deduced from the nucleotide sequence of individual genes. -

Ionization, Dissociation, and the Investigation of Structure in Small Molecular Models to Develop a Broader Understanding of Gas Phase Ion Chemistry
Ionization, dissociation, and the investigation of structure in small molecular models to develop a broader understanding of gas phase ion chemistry DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Matthew Bernier Graduate Program in Chemistry The Ohio State University 2014 Dissertation Committee: Professor Vicki Wysocki, Advisor Professor Terry Gustafson Professor Abraham Badu-Tawiah Copyright by Matthew Bernier 2014 Abstract This dissertation focuses on molecular systems in the low-mass range to determine how chemical and structural changes can affect subsequent fragmentation chemistry and protonation site. Each system was investigated using MS analysis and gas-phase ion structural techniques selected from tandem MS (MSMS), hydrogen-deuterium exchange (HDX), ion-mobility (IM), and action infra-red multi-photon dissociation (IRMPD). In Chapter 3, the non-standard amino acid gamma-aminobutyric acid (GABA) was placed into a peptide system to test a mechanism which explained the lack of a3 ions in standard peptide fragmentations. GABA extends the peptide backbone by two methylene units and its insertion into the second position of larger peptides increases the intensity of a3 ion. Using MSMS, it was found that this was a result of blocking common favorable fragmentation pathways. The results demonstrated the use of modified peptides for revealing reasons for how peptides fragment. Chapter 4 focused on a unique set of non-canonical amino acids and their ability to affect the trans/cis nature of adjacent amide bonds in peptide sequences. In solution, 4- R-Flouroproline (R-Flp) is found to favor the trans peptide bond and 4-S-fluoroproline (S- flp) favors the cis bond. -

Drugs and Acid Dissociation Constants Ionisation of Drug Molecules Most Drugs Ionise in Aqueous Solution.1 They Are Weak Acids Or Weak Bases
Drugs and acid dissociation constants Ionisation of drug molecules Most drugs ionise in aqueous solution.1 They are weak acids or weak bases. Those that are weak acids ionise in water to give acidic solutions while those that are weak bases ionise to give basic solutions. Drug molecules that are weak acids Drug molecules that are weak bases where, HA = acid (the drug molecule) where, B = base (the drug molecule) H2O = base H2O = acid A− = conjugate base (the drug anion) OH− = conjugate base (the drug anion) + + H3O = conjugate acid BH = conjugate acid Acid dissociation constant, Ka For a drug molecule that is a weak acid The equilibrium constant for this ionisation is given by the equation + − where [H3O ], [A ], [HA] and [H2O] are the concentrations at equilibrium. In a dilute solution the concentration of water is to all intents and purposes constant. So the equation is simplified to: where Ka is the acid dissociation constant for the weak acid + + Also, H3O is often written simply as H and the equation for Ka is usually written as: Values for Ka are extremely small and, therefore, pKa values are given (similar to the reason pH is used rather than [H+]. The relationship between pKa and pH is given by the Henderson–Hasselbalch equation: or This relationship is important when determining pKa values from pH measurements. Base dissociation constant, Kb For a drug molecule that is a weak base: 1 Ionisation of drug molecules. 1 Following the same logic as for deriving Ka, base dissociation constant, Kb, is given by: and Ionisation of water Water ionises very slightly. -

Introduction to Chemistry
Introduction to Chemistry Author: Tracy Poulsen Digital Proofer Supported by CK-12 Foundation CK-12 Foundation is a non-profit organization with a mission to reduce the cost of textbook Introduction to Chem... materials for the K-12 market both in the U.S. and worldwide. Using an open-content, web-based Authored by Tracy Poulsen collaborative model termed the “FlexBook,” CK-12 intends to pioneer the generation and 8.5" x 11.0" (21.59 x 27.94 cm) distribution of high-quality educational content that will serve both as core text as well as provide Black & White on White paper an adaptive environment for learning. 250 pages ISBN-13: 9781478298601 Copyright © 2010, CK-12 Foundation, www.ck12.org ISBN-10: 147829860X Except as otherwise noted, all CK-12 Content (including CK-12 Curriculum Material) is made Please carefully review your Digital Proof download for formatting, available to Users in accordance with the Creative Commons Attribution/Non-Commercial/Share grammar, and design issues that may need to be corrected. Alike 3.0 Unported (CC-by-NC-SA) License (http://creativecommons.org/licenses/by-nc- sa/3.0/), as amended and updated by Creative Commons from time to time (the “CC License”), We recommend that you review your book three times, with each time focusing on a different aspect. which is incorporated herein by this reference. Specific details can be found at http://about.ck12.org/terms. Check the format, including headers, footers, page 1 numbers, spacing, table of contents, and index. 2 Review any images or graphics and captions if applicable. -
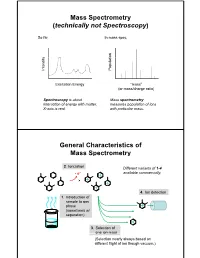
Mass Spectrometry (Technically Not Spectroscopy)
Mass Spectrometry (technically not Spectroscopy) So far, In mass spec, on y Populati Intensit Excitation Energy “mass” (or mass/charge ratio) Spectroscopy is about Mass spectrometry interaction of energy with matter. measures population of ions X-axis is real. with particular mass. General Characteristics of Mass Spectrometry 2. Ionization Different variants of 1-4 -e- available commercially. 4. Ion detection 1. Introduction of sample to gas phase (sometimes w/ separation) 3. Selection of one ion mass (Selection nearly always based on different flight of ion though vacuum.) General Components of a Mass Spectrometer Lots of choices, which can be mixed and matched. direct injection The Mass Spectrum fragment “daughter” ions M+ “parent” mass Sample Introduction: Direc t Inser tion Prob e If sample is a liquid, sample can also be injected directly into ionization region. If sample isn’t pure, get multiple parents (that can’t be distinguished from fragments). Capillary Column Introduction Continous source of molecules to spectrometer. detector column (including GC, LC, chiral, size exclusion) • Signal intensity depends on both amount of molecule and ionization efficiency • To use quantitatively, must calibrate peaks with respect eltilution time ttlitotal ion curren t to quantity eluted (TIC) over time Capillary Column Introduction Easy to interface with gas or liquid chromatography. TIC trace elution time time averaged time averaged mass spectrum mass spectrum Methods of Ionization: Electron Ionization (EI) 1 - + - 1 M + e (kV energy) M + 2e Fragmentation in Electron Ionization daughter ion (observed in spectrum) neutral fragment (not observed) excited parent at electron at electron energy of energy of 15 eV 70 e V Lower electron energy yields less fragmentation, but also less signal. -

An Introduction to Mass Spectrometry
An Introduction to Mass Spectrometry by Scott E. Van Bramer Widener University Department of Chemistry One University Place Chester, PA 19013 [email protected] http://science.widener.edu/~svanbram revised: September 2, 1998 © Copyright 1997 TABLE OF CONTENTS INTRODUCTION ........................................................... 4 SAMPLE INTRODUCTION ....................................................5 Direct Vapor Inlet .......................................................5 Gas Chromatography.....................................................5 Liquid Chromatography...................................................6 Direct Insertion Probe ....................................................6 Direct Ionization of Sample ................................................6 IONIZATION TECHNIQUES...................................................6 Electron Ionization .......................................................7 Chemical Ionization ..................................................... 9 Fast Atom Bombardment and Secondary Ion Mass Spectrometry .................10 Atmospheric Pressure Ionization and Electrospray Ionization ....................11 Matrix Assisted Laser Desorption/Ionization ................................ 13 Other Ionization Methods ................................................13 Self-Test #1 ...........................................................14 MASS ANALYZERS .........................................................14 Quadrupole ............................................................15 -

Methods of Ion Generation
Chem. Rev. 2001, 101, 361−375 361 Methods of Ion Generation Marvin L. Vestal PE Biosystems, Framingham, Massachusetts 01701 Received May 24, 2000 Contents I. Introduction 361 II. Atomic Ions 362 A. Thermal Ionization 362 B. Spark Source 362 C. Plasma Sources 362 D. Glow Discharge 362 E. Inductively Coupled Plasma (ICP) 363 III. Molecular Ions from Volatile Samples. 364 A. Electron Ionization (EI) 364 B. Chemical Ionization (CI) 365 C. Photoionization (PI) 367 D. Field Ionization (FI) 367 IV. Molecular Ions from Nonvolatile Samples 367 Marvin L. Vestal received his B.S. and M.S. degrees, 1958 and 1960, A. Spray Techniques 367 respectively, in Engineering Sciences from Purdue Univesity, Layfayette, IN. In 1975 he received his Ph.D. degree in Chemical Physics from the B. Electrospray 367 University of Utah, Salt Lake City. From 1958 to 1960 he was a Scientist C. Desorption from Surfaces 369 at Johnston Laboratories, Inc., in Layfayette, IN. From 1960 to 1967 he D. High-Energy Particle Impact 369 became Senior Scientist at Johnston Laboratories, Inc., in Baltimore, MD. E. Low-Energy Particle Impact 370 From 1960 to 1962 he was a Graduate Student in the Department of Physics at John Hopkins University. From 1967 to 1970 he was Vice F. Low-Energy Impact with Liquid Surfaces 371 President at Scientific Research Instruments, Corp. in Baltimore, MD. From G. Flow FAB 371 1970 to 1975 he was a Graduate Student and Research Instructor at the H. Laser Ionization−MALDI 371 University of Utah, Salt Lake City. From 1976 to 1981 he became I. -

Selective Chemical Ionization of Nitrogen and Sulfur Heterocycles in Petroleum Fractions by Ion Trap Mass Spectrometry
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Selective Chemical Ionization of Nitrogen and Sulfur Heterocycles in Petroleum Fractions by Ion Trap Mass Spectrometry C. S. Creaser*, F. Krokos, and K. E. O’Neill Shod of Chemical Sciences, University of East Anglia, Norwich, United Kingdom M. J. C. Smith and P. G. McDowell BP Research, Sunbury-on-Thames, Middlesex, United Kingdom A procedure is reported for the selective ammonia chemical ionization of some nitrogen and sulfur heterocycles in petroleum fractions using ion trap mass spectrometry (ITMS). The ion trap scan routine is designed to optimize the population of ammonium reagent ions and eject from the trap (by radio frequency/direct current isolation) electron ionization products formed during reagent ion formation prior to ionization of the sample. The ITMS procedure is compared with standard ion trap detector and conventional quadrupole ammonia chemi- cal ionization for the determination of nitrogen and sulfur heterocycles in gas oil and kerosine samples. Greatly enhanced selectivity is shown for the ITMS procedure by sup res- sion of competing charge-exchange processes. (1 Am Sot Mass Spectrum 1993, 4, 322-326 ‘; he removal of organic compounds containing reagent ion NH;, is formed via the reactions [5] nitrogen and sulfur is an important part of the T petroleum-refining process 111. Their presence in NH, ‘1 NH;’ (1) petroleum distillates results in the formation of envi- NH, f NH:‘+ NH; + NH; (2) ronmental pollutants (SO,, NO,) during combustion, and their reactivity can lead to catalyst poisoning Whereas NH: is unreactive toward a wide range of in refinery processes.