Infrared Laser Desorption/Ionization Mass Spectrometry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

An Application of the Theory of Laser to Nitrogen Laser Pumped Dye Laser
SD9900039 AN APPLICATION OF THE THEORY OF LASER TO NITROGEN LASER PUMPED DYE LASER FATIMA AHMED OSMAN A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science in Physics. UNIVERSITY OF KHARTOUM FACULTY OF SCIENCE DEPARTMENT OF PHYSICS MARCH 1998 \ 3 0-44 In this thesis we gave a general discussion on lasers, reviewing some of are properties, types and applications. We also conducted an experiment where we obtained a dye laser pumped by nitrogen laser with a wave length of 337.1 nm and a power of 5 Mw. It was noticed that the produced radiation possesses ^ characteristic^ different from those of other types of laser. This' characteristics determine^ the tunability i.e. the possibility of choosing the appropriately required wave-length of radiation for various applications. DEDICATION TO MY BELOVED PARENTS AND MY SISTER NADI A ACKNOWLEDGEMENTS I would like to express my deep gratitude to my supervisor Dr. AH El Tahir Sharaf El-Din, for his continuous support and guidance. I am also grateful to Dr. Maui Hammed Shaded, for encouragement, and advice in using the computer. Thanks also go to Ustaz Akram Yousif Ibrahim for helping me while conducting the experimental part of the thesis, and to Ustaz Abaker Ali Abdalla, for advising me in several respects. I also thank my teachers in the Physics Department, of the Faculty of Science, University of Khartoum and my colleagues and co- workers at laser laboratory whose support and encouragement me created the right atmosphere of research for me. Finally I would like to thank my brother Salah Ahmed Osman, Mr. -

Theoretical Calculations for Electron Impact Ionization of Atoms and Molecules
Scholars' Mine Doctoral Dissertations Student Theses and Dissertations Fall 2017 Theoretical calculations for electron impact ionization of atoms and molecules Sadek Mohamed Fituri Amami Follow this and additional works at: https://scholarsmine.mst.edu/doctoral_dissertations Part of the Atomic, Molecular and Optical Physics Commons Department: Physics Recommended Citation Amami, Sadek Mohamed Fituri, "Theoretical calculations for electron impact ionization of atoms and molecules" (2017). Doctoral Dissertations. 2617. https://scholarsmine.mst.edu/doctoral_dissertations/2617 This thesis is brought to you by Scholars' Mine, a service of the Missouri S&T Library and Learning Resources. This work is protected by U. S. Copyright Law. Unauthorized use including reproduction for redistribution requires the permission of the copyright holder. For more information, please contact [email protected]. THEORETICAL CALCULATIONS FOR ELECTRON IMPACT IONIZATION OF ATOMS AND MOLECULES by SADEK MOHAMED FITURI AMAMI A DISSERTATION Presented to the Graduate Faculty of the MISSOURI UNIVERSITY OF SCIENCE AND TECHNOLOGY In Partial Fulfillment of the Requirements for the Degree DOCTOR OF PHILOSOPHY in PHYSICS 2017 Approved by Don H. Madison, Advisor Jerry L. Peacher Michael Schulz Daniel Fischer Gregory Gelles Copyright 2017 SADEK MOHAMED FITURI AMAMI All Rights Reserved iii PUBLICATION DISSERTATION OPTION This dissertation consists of the following ten articles which have been published as follows: Paper I: Pages 31-54 have been published by Phys Rev A.91.032707 (March 2015). Paper II: Pages 55-78 have been published by Phys Rev A.90.012704 (July 2014). Paper III: Pages 79-93 have been published by J. Phys. B: At. Mol. Opt. Phys. 49 185202 (September 2016). -

Development and Characterization of an Atmospheric Pressure Ionization Source Matrix-Assisted Laser Desorption Electrospray Ioni
ABSTRACT SAMPSON, JASON SCOTT. Development and Characterization of an Atmospheric Pressure Ionization Source Matrix-Assisted Laser Desorption Electrospray Ionization Coupled to Fourier Transform Ion Cyclotron Resonance Mass Spectrometry for Analysis of Biological Macromolecules. (Under the direction of David Charles Muddiman). The field of mass spectrometry has grown tremendously over the past few decades due in large part to the continued application to new and interesting areas of exploration. The advent of soft ionization sources such as electrospray ionization and matrix-assisted laser desorption/ionization has dramatically increased the applications of mass spectrometry; in particular, analysis of complex biological samples. Electrospray ionization has demonstrated the capability to generate multiply-charged ions which increases the amount of information garnered from each analysis. Matrix-assisted laser desorption/ionization has demonstrated complex sample analysis requiring minimal sample preparation, which results in high throughput. As a result of these findings, there has been tremendous growth in the development of new ionization source technology in recent years for reducing sample preparation required prior to analysis for high throughput sample analysis and the generation of multiply-charged ions. Demonstrated herein is the development and characterization of an atmospheric pressure ionization source called matrix-assisted laser desorption electrospray ionization (MALDESI). MALDESI is a hybrid combination of MALDI and ESI which utilizes laser desorption with electrospray postionization for the generation of multiply-charged ions. Multiply-charged ions are of particular importance when using Fourier transform mass spectrometry, due to the increase in resolving power and mass accuracy with increasing charge on the molecule. Positive identification of biological macromolecules is demonstrated utilizing top-down characterization of intact polypeptides as well as high mass accuracy utilizing internal calibration. -

With Femtosecond Lasers, 572-74, 573/ Hydrodynamic Motion
Index Ablation Central hot spark ignition, by direct drive of ceramics, 216-21 implosion, 380-83 dielectric function during, 192-93, 193/ Central ignition scheme, 377/ 378 with femtosecond lasers, 572-74, 573/ Ceramics, ultrashort laser pulse interactions hydrodynamic motion role in, 2, 5-6 with, 216-21,217/218/ initial temperature at threshold of, 3 Coherent anti-Stokes Raman scattering measurement of dynamics of, 190, 191/ (CARS), 198, 206-8, 207/ of polymers, 225-26, 225/ 226/ 281-95 Coherent control of polymers with EUV laser, 535^0, 536/ raman spectroscopy, 206-8, 207/ 537/ 538/ 538<, 539/ of ultrashort laser pulse, 203^, 203/ spectral control of, 204-5, 205/ 206/ Collisional ionization, of dielectrics, 26-27 stages of, 68 69, 69/ Cosine model, of laser ionization, 110-14, Ablative cleaning, 37 110/115/ Ablative laser propulsion, 408-11, 409/ Coulomb explosion (CE) Artworks. See Painted artworks criterion for, 20-21, 21/ Asymptotic density profile, 12, 13/ features of, 18 from ultrashort laser pulse, 17-32 Band structure, 99 experimental evidence for, 18-20 Biological scaffold structures, from Crater ORMOCER®s, 149-51, 150/ 151/ formation of, 7-8, If Boltzmann plot, of plume, 77, 77/ hydrodynamic motion formation of, 4 Brillouin zone (BZ), of laser ionization, Crystal expansion, ultrashort laser pulse for, 100-108, 101/ 108/ 110 1-15 Bubble formation conclusion of, 14-15 calculation of, 254-57 hydrodynamic motion characteristics, 4—6 cavitation bubble dynamics, 262-67, 263/ hydrodynamic simulation, 11-14 264/ 265/ interference during, 9-11,10/ pulse trains with energies above threshold introduction to, 1-3 for, 271-72 shell and crater formation, 7-8, If pulse trains with energies below threshold for, 269-71,269/ Desorption ionization on silicon (DIOS) stress-induced, 254—67 MALDIv., 518-19, 518/521/ threshold for, 261-62 asSLDI, 506-7, 515-19 BZ. -

Electron Ionization
Chapter 6 Chapter 6 Electron Ionization I. Introduction ......................................................................................................317 II. Ionization Process............................................................................................317 III. Strategy for Data Interpretation......................................................................321 1. Assumptions 2. The Ionization Process IV. Types of Fragmentation Pathways.................................................................328 1. Sigma-Bond Cleavage 2. Homolytic or Radical-Site-Driven Cleavage 3. Heterolytic or Charge-Site-Driven Cleavage 4. Rearrangements A. Hydrogen-Shift Rearrangements B. Hydride-Shift Rearrangements V. Representative Fragmentations (Spectra) of Classes of Compounds.......... 344 1. Hydrocarbons A. Saturated Hydrocarbons 1) Straight-Chain Hydrocarbons 2) Branched Hydrocarbons 3) Cyclic Hydrocarbons B. Unsaturated C. Aromatic 2. Alkyl Halides 3. Oxygen-Containing Compounds A. Aliphatic Alcohols B. Aliphatic Ethers C. Aromatic Alcohols D. Cyclic Ethers E. Ketones and Aldehydes F. Aliphatic Acids and Esters G. Aromatic Acids and Esters 4. Nitrogen-Containing Compounds A. Aliphatic Amines B. Aromatic Compounds Containing Atoms of Nitrogen C. Heterocyclic Nitrogen-Containing Compounds D. Nitro Compounds E. Concluding Remarks on the Mass Spectra of Nitrogen-Containing Compounds 5. Multiple Heteroatoms or Heteroatoms and a Double Bond 6. Trimethylsilyl Derivative 7. Determining the Location of Double Bonds VI. Library -

Modern Mass Spectrometry
Modern Mass Spectrometry MacMillan Group Meeting 2005 Sandra Lee Key References: E. Uggerud, S. Petrie, D. K. Bohme, F. Turecek, D. Schröder, H. Schwarz, D. Plattner, T. Wyttenbach, M. T. Bowers, P. B. Armentrout, S. A. Truger, T. Junker, G. Suizdak, Mark Brönstrup. Topics in Current Chemistry: Modern Mass Spectroscopy, pp. 1-302, 225. Springer-Verlag, Berlin, 2003. Current Topics in Organic Chemistry 2003, 15, 1503-1624 1 The Basics of Mass Spectroscopy ! Purpose Mass spectrometers use the difference in mass-to-charge ratio (m/z) of ionized atoms or molecules to separate them. Therefore, mass spectroscopy allows quantitation of atoms or molecules and provides structural information by the identification of distinctive fragmentation patterns. The general operation of a mass spectrometer is: "1. " create gas-phase ions "2. " separate the ions in space or time based on their mass-to-charge ratio "3. " measure the quantity of ions of each mass-to-charge ratio Ionization sources ! Instrumentation Chemical Ionisation (CI) Atmospheric Pressure CI!(APCI) Electron Impact!(EI) Electrospray Ionization!(ESI) SORTING DETECTION IONIZATION OF IONS OF IONS Fast Atom Bombardment (FAB) Field Desorption/Field Ionisation (FD/FI) Matrix Assisted Laser Desorption gaseous mass ion Ionisation!(MALDI) ion source analyzer transducer Thermospray Ionisation (TI) Analyzers quadrupoles vacuum signal Time-of-Flight (TOF) pump processor magnetic sectors 10-5– 10-8 torr Fourier transform and quadrupole ion traps inlet Detectors mass electron multiplier spectrum Faraday cup Ionization Sources: Classical Methods ! Electron Impact Ionization A beam of electrons passes through a gas-phase sample and collides with neutral analyte molcules (M) to produce a positively charged ion or a fragment ion. -
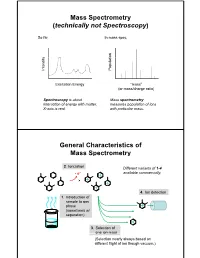
Mass Spectrometry (Technically Not Spectroscopy)
Mass Spectrometry (technically not Spectroscopy) So far, In mass spec, on y Populati Intensit Excitation Energy “mass” (or mass/charge ratio) Spectroscopy is about Mass spectrometry interaction of energy with matter. measures population of ions X-axis is real. with particular mass. General Characteristics of Mass Spectrometry 2. Ionization Different variants of 1-4 -e- available commercially. 4. Ion detection 1. Introduction of sample to gas phase (sometimes w/ separation) 3. Selection of one ion mass (Selection nearly always based on different flight of ion though vacuum.) General Components of a Mass Spectrometer Lots of choices, which can be mixed and matched. direct injection The Mass Spectrum fragment “daughter” ions M+ “parent” mass Sample Introduction: Direc t Inser tion Prob e If sample is a liquid, sample can also be injected directly into ionization region. If sample isn’t pure, get multiple parents (that can’t be distinguished from fragments). Capillary Column Introduction Continous source of molecules to spectrometer. detector column (including GC, LC, chiral, size exclusion) • Signal intensity depends on both amount of molecule and ionization efficiency • To use quantitatively, must calibrate peaks with respect eltilution time ttlitotal ion curren t to quantity eluted (TIC) over time Capillary Column Introduction Easy to interface with gas or liquid chromatography. TIC trace elution time time averaged time averaged mass spectrum mass spectrum Methods of Ionization: Electron Ionization (EI) 1 - + - 1 M + e (kV energy) M + 2e Fragmentation in Electron Ionization daughter ion (observed in spectrum) neutral fragment (not observed) excited parent at electron at electron energy of energy of 15 eV 70 e V Lower electron energy yields less fragmentation, but also less signal. -

Nitrogen Laser Emissions of Short and Long Durations Generated in Air Mladen M
IEEE TRANSACTIONS ON PLASMA SCIENCE, VOL. 48, NO. 3, MARCH 2020 647 Nitrogen Laser Emissions of Short and Long Durations Generated in Air Mladen M. Kekez and David M. Villeneuve Abstract— This article describes two sets of experiments. pressure, the excitation of the N2 molecules, and consequently An eight-stage, pulse-forming network (PFN) Marx generator the laser emission would yield only short duration light pulses, of 50- internal impedance produces a “square”-shaped voltage from one nanosecond down to some tens of picoseconds. waveform and induces multiphoton ionization, excitation of molecules and ions to generate laser pulses of long (<3 μs) and From the research and development on CO2 lasers, it was short (∼10 ns) durations in air, when the principal laser line of learned that the delay in the transition from the glow-like the N laser at 337.1 nm was observed. The visible blue laser discharge to a fully developed spark channel is possible, when 2 + lines coming from the first negative band of N2 ions due to the the characteristic impedance of the driver is of relatively large 2 + → 2 + B u X g transition at 391.4 and 427.8 nm were studied. value (>10 ). The wideband (300–800 nm) Schott BG 39 interference filter was To explore the importance, the nine-stage Marx generator also employed to examine whether some other lines participate in the emission being generated. The scaling law was derived of 18- internal impedance was used in [5]. The electro- describing the attenuation of theemissionversusthedistance magnetic radiation pulses were observed both at the single for long-duration pulses (<3 μs) at 337.1 nm. -

An Organic Chemist's Guide to Electrospray Mass Spectrometric
molecules Review An Organic Chemist’s Guide to Electrospray Mass Spectrometric Structure Elucidation Arnold Steckel 1 and Gitta Schlosser 2,* 1 Hevesy György PhD School of Chemistry, ELTE Eötvös Loránd University, Pázmány Péter sétány 1/A, 1117 Budapest, Hungary; [email protected] 2 Department of Analytical Chemistry, ELTE Eötvös Loránd University, Pázmány Péter sétány 1/A, 1117 Budapest, Hungary * Correspondence: [email protected] Received: 16 January 2019; Accepted: 8 February 2019; Published: 10 February 2019 Abstract: Tandem mass spectrometry is an important tool for structure elucidation of natural and synthetic organic products. Fragmentation of odd electron ions (OE+) generated by electron ionization (EI) was extensively studied in the last few decades, however there are only a few systematic reviews available concerning the fragmentation of even-electron ions (EE+/EE−) produced by the currently most common ionization techniques, electrospray ionization (ESI) and atmospheric pressure chemical ionization (APCI). This review summarizes the most important features of tandem mass spectra generated by collision-induced dissociation fragmentation and presents didactic examples for the unexperienced users. Keywords: tandem mass spectrometry; MS/MS fragmentation; collision-induced dissociation; CID; ESI; structure elucidation 1. Introduction Electron ionization (EI), a hard ionization technique, is the method of choice for analyses of small (<1000 Da), nonpolar, volatile compounds. As its name implies, the technique involves ionization by electrons with ~70 eV energy. This energy is high enough to yield very reproducible mass spectra with a large number of fragments. However, these spectra frequently lack the radical type molecular ions (M+) due to the high internal energy transferred to the precursors [1]. -

Methods of Ion Generation
Chem. Rev. 2001, 101, 361−375 361 Methods of Ion Generation Marvin L. Vestal PE Biosystems, Framingham, Massachusetts 01701 Received May 24, 2000 Contents I. Introduction 361 II. Atomic Ions 362 A. Thermal Ionization 362 B. Spark Source 362 C. Plasma Sources 362 D. Glow Discharge 362 E. Inductively Coupled Plasma (ICP) 363 III. Molecular Ions from Volatile Samples. 364 A. Electron Ionization (EI) 364 B. Chemical Ionization (CI) 365 C. Photoionization (PI) 367 D. Field Ionization (FI) 367 IV. Molecular Ions from Nonvolatile Samples 367 Marvin L. Vestal received his B.S. and M.S. degrees, 1958 and 1960, A. Spray Techniques 367 respectively, in Engineering Sciences from Purdue Univesity, Layfayette, IN. In 1975 he received his Ph.D. degree in Chemical Physics from the B. Electrospray 367 University of Utah, Salt Lake City. From 1958 to 1960 he was a Scientist C. Desorption from Surfaces 369 at Johnston Laboratories, Inc., in Layfayette, IN. From 1960 to 1967 he D. High-Energy Particle Impact 369 became Senior Scientist at Johnston Laboratories, Inc., in Baltimore, MD. E. Low-Energy Particle Impact 370 From 1960 to 1962 he was a Graduate Student in the Department of Physics at John Hopkins University. From 1967 to 1970 he was Vice F. Low-Energy Impact with Liquid Surfaces 371 President at Scientific Research Instruments, Corp. in Baltimore, MD. From G. Flow FAB 371 1970 to 1975 he was a Graduate Student and Research Instructor at the H. Laser Ionization−MALDI 371 University of Utah, Salt Lake City. From 1976 to 1981 he became I. -

Laser Threshold
Main Requirements of the Laser • Optical Resonator Cavity • Laser Gain Medium of 2, 3 or 4 level types in the Cavity • Sufficient means of Excitation (called pumping) eg. light, current, chemical reaction • Population Inversion in the Gain Medium due to pumping Laser Types • Two main types depending on time operation • Continuous Wave (CW) • Pulsed operation • Pulsed is easier, CW more useful Optical Resonator Cavity • In laser want to confine light: have it bounce back and forth • Then it will gain most energy from gain medium • Need several passes to obtain maximum energy from gain medium • Confine light between two mirrors (Resonator Cavity) Also called Fabry Perot Etalon • Have mirror (M1) at back end highly reflective • Front end (M2) not fully transparent • Place pumped medium between two mirrors: in a resonator • Needs very careful alignment of the mirrors (arc seconds) • Only small error and cavity will not resonate • Curved mirror will focus beam approximately at radius • However is the resonator stable? • Stability given by g parameters: g1 back mirror, g2 front mirror: L gi = 1 − ri • For two mirrors resonator stable if 0 < g1g2 < 1 • Unstable if g1g2 < 0 g1g2 > 1 • At the boundary (g1g2 = 0 or 1) marginally stable Stability of Different Resonators • If plot g1 vs g2 and 0 < g1g2 < 1 then get a stability plot • Now convert the g’s also into the mirror shapes Polarization of Light • Polarization is where the E and B fields aligned in one direction • All the light has E field in same direction • Black Body light is not polarized -

Theoretical Model of TEA Nitrogen Laser Excited by Electric Discharge. Part 3
Optica Applicata, Vol. X X III, No. 4, 1993 Theoretical model of TEA nitrogen laser excited by electric discharge. Part 3. Construction and the preliminary results of the experimental setup examination J. Makuchowski, L. Pokora Laser Technics Centre, uL Kasprzaka 29/31, 01-234 Warszawa, Poland. A construction of two utilizable setups of nitrogen laser of the TEA-type produced on the basis of the experimental model described in Parts 1 and 2 [1], [2] of this work has been described. The realized models of nitrogen lasers were subjected to examinations. The preliminary results of these examinations were compared with the results of the respective theoretical calculations. High degree of consistence of the measurement results with those of the corresponding simulation calculations was achieved. 1. Introduction Due to attractive properties of radiation emitted by nitrogen lasers, in this field research in this field is being carried out almost continuously by different centres of science and technology. In a series of articles devoted to construction of nitrogen lasers pumped with transversal electric discharge [3] — [15], the energies and durations of pulses are reported together with some parameters of electric circuits (usually the energy accumulated in the capacitors and the supply voltage). There are very few descriptions of the scheme of electric circuit and details of the chamber structure of nitrogen laser are rarely provided. As was mentioned in the previous parts of this work [1], [2], the constructions described in the literature are difficult to reproduce so as to obtain either optimal or required parameters (or characteristics). In practice, two regions of nitrogen laser parameters are useful, ie., those for lasers generating nano- or subnanosecond pulses of radiation of high peak power, or those for lasers of relatively high energy generated in longer pulses.