The Structural Basis for Pyrophosphatase Catalysis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Identification and Partial Characterization of a Family of Putative Palmitoyltransferases in Dictyostelium Discoideum Brent Elliot Wells
The University of Maine DigitalCommons@UMaine Electronic Theses and Dissertations Fogler Library 2003 Identification and Partial Characterization of a Family of Putative Palmitoyltransferases in Dictyostelium Discoideum Brent Elliot Wells Follow this and additional works at: http://digitalcommons.library.umaine.edu/etd Part of the Biochemistry Commons Recommended Citation Wells, Brent Elliot, "Identification and Partial Characterization of a Family of Putative Palmitoyltransferases in Dictyostelium Discoideum" (2003). Electronic Theses and Dissertations. 301. http://digitalcommons.library.umaine.edu/etd/301 This Open-Access Thesis is brought to you for free and open access by DigitalCommons@UMaine. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of DigitalCommons@UMaine. IDENTIFICATION AND PARTIAL CHARACTERIZATION OF A FAMILY OF PUTATIVE PALMITOYLTRANSFERASES IN DICTYOSTELIUM DISCOIDEUM BY Brent Elliott Wells B.S. University of Utah, 1994 A THESIS Submitted in Partial Fulfillment of the Requirements for the Degree of Master of Science (in Biochemistry) The Graduate School The University of Maine December, 2003 Advisory Committee: Robert E. Gundersen, Associate Professor of Biochemistry, Advisor Keith W. Hutchison, Professor of Biochemistry Mary Rumpho-Kennedy, Professor of Biochemistry LIBRARY RIGHTS STATEMENT In presenting this thesis in partial fulfillment of the requirements for an advanced degree at The University of Maine, I agree that the Library shall make it freely available for inspection. I further agree that permission for "fair use" copying of this thesis for scholarly purposes may be granted by the Librarian. It is understood that any copying or publication of this thesis for financial gain shall not be allowed without my written permission. -

Inorganic Pyrophosphatase Is a Component of the Drosophila Nucleosome Remodeling Factor Complex
Downloaded from genesdev.cshlp.org on October 3, 2021 - Published by Cold Spring Harbor Laboratory Press Inorganic pyrophosphatase is a component of the Drosophila nucleosome remodeling factor complex David A. Gdula, Raphael Sandaltzopoulos, Toshio Tsukiyama,1 Vincent Ossipow, and Carl Wu2 Laboratory of Molecular Cell Biology, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892-4255 USA The Drosophila nucleosome remodeling factor (NURF) is a protein complex consisting of four polypeptides that facilitates the perturbation of chromatin structure in vitro in an ATP-dependent manner. The 140-kD NURF subunit, imitation switch (ISWI), is related to the SWI2/SNF2 ATPase. Another subunit, NURF-55, is a 55-kD WD repeat protein homologous to the human retinoblastoma-associated protein RbAp48. Here, we report the cloning and characterization of the smallest (38 kD) component of NURF. NURF-38 is strikingly homologous to known inorganic pyrophosphatases. Both recombinant NURF-38 alone and the purified NURF complex are shown to have inorganic pyrophosphatase activity. Inhibition of the pyrophosphatase activity of NURF with sodium fluoride has no significant effect on chromatin remodeling, indicating that these two activities may be biochemically uncoupled. Our results suggest that NURF-38 may serve a structural or regulatory role in the complex. Alternatively, because accumulation of unhydrolyzed pyrophosphate during nucleotide incorporation inhibits polymerization, NURF may also have been adapted to deliver pyrophosphatase -
Type of the Paper (Article
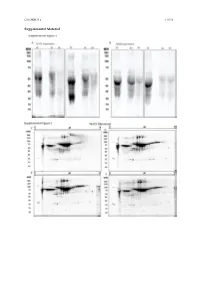
Cells 2020, 9, x 1 of 19 Supplemental Material Cells 2020, 9, x 2 of 19 Figure S1. Secretome enrichment: protocol optimization. 1D SDS-PAGE documentation of washing steps: Culture medium was substituted with FCS-free medium, which was changed every 2 h. The supernatants were then collected, and the proteins isolated and separated in 1D SDS-PAGE ((A) TK173 and (B) TK188). Proteins were stained with Flamingo fluorescent gel stain. Two-dimensional pattern of the proteins isolated from supernatant of TK173, (C) 2 h, (D) 4 h, (E) 6 h, and (F) 8 h after changing to FCS-free medium. (G) Cell secretome collected 24 h after elimination of the contaminating FCS-proteins with different washing steps. Proteins were stained with Flamingo fluorescent gel stain. Cells 2020, 9, x 3 of 19 Figure S2. 2-DE reference maps of secretomes; 150 μg proteins were loaded on an 11 cm IPG strip with a linear pH gradient PI 5–8 for IEF; 12% SDS-polyacrylamide gels were used for the second dimension. Proteins were stained with Flamingo fluorescent gel stain. Identified spots were assigned a number corresponding to that in their table. 2-DE maps from secretome of (A) TK173 control and (B) TGFβ1- treated ones. The 2-DE patterns revealed an alteration of secretome in stimulated TK173. Secretome patterns from TK173 treated with (C) ANG II and (D) PDGF. Cells 2020, 9, x 4 of 19 A Figure S3. Classification of the differentially expressed proteins upon ANG II, TGFβ1, or PDGF treatment in TK173. (A) Bar charts of the cellular component analyzed by STRAP biological function analysis in which the identified proteins from all treatments in both cell types are involved. -

In Vivo Mapping of a GPCR Interactome Using Knockin Mice
In vivo mapping of a GPCR interactome using knockin mice Jade Degrandmaisona,b,c,d,e,1, Khaled Abdallahb,c,d,1, Véronique Blaisb,c,d, Samuel Géniera,c,d, Marie-Pier Lalumièrea,c,d, Francis Bergeronb,c,d,e, Catherine M. Cahillf,g,h, Jim Boulterf,g,h, Christine L. Lavoieb,c,d,i, Jean-Luc Parenta,c,d,i,2, and Louis Gendronb,c,d,i,j,k,2 aDépartement de Médecine, Université de Sherbrooke, Sherbrooke, QC J1H 5N4, Canada; bDépartement de Pharmacologie–Physiologie, Université de Sherbrooke, Sherbrooke, QC J1H 5N4, Canada; cFaculté de Médecine et des Sciences de la Santé, Université de Sherbrooke, Sherbrooke, QC J1H 5N4, Canada; dCentre de Recherche du Centre Hospitalier Universitaire de Sherbrooke, Sherbrooke, QC J1H 5N4, Canada; eQuebec Network of Junior Pain Investigators, Sherbrooke, QC J1H 5N4, Canada; fDepartment of Psychiatry and Biobehavioral Sciences, University of California, Los Angeles, CA 90095; gSemel Institute for Neuroscience and Human Behavior, University of California, Los Angeles, CA 90095; hShirley and Stefan Hatos Center for Neuropharmacology, University of California, Los Angeles, CA 90095; iInstitut de Pharmacologie de Sherbrooke, Sherbrooke, QC J1H 5N4, Canada; jDépartement d’Anesthésiologie, Université de Sherbrooke, Sherbrooke, QC J1H 5N4, Canada; and kQuebec Pain Research Network, Sherbrooke, QC J1H 5N4, Canada Edited by Brian K. Kobilka, Stanford University School of Medicine, Stanford, CA, and approved April 9, 2020 (received for review October 16, 2019) With over 30% of current medications targeting this family of attenuates pain hypersensitivities in several chronic pain models proteins, G-protein–coupled receptors (GPCRs) remain invaluable including neuropathic, inflammatory, diabetic, and cancer pain therapeutic targets. -

ALLOSTERIC REGULATION of GS on AGONIST, ANTAGONIST and INVERSE AGONIST BINDING to the Β2ar by Gisselle A. Vélez Ruiz a Dissert
ALLOSTERIC REGULATION OF GS ON AGONIST, ANTAGONIST AND INVERSE AGONIST BINDING TO THE β2AR by Gisselle A. Vélez Ruiz A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Cellular and Molecular Biology) in the University of Michigan 2011 Doctoral Committee: Associate Professor Roger K. Sunahara, Chair Professor Richard R. Neubig Professor Michael D. Uhler Associate Professor Martin Myers Associate Professor John J.G. Tesmer To my family especially my mom and siblings, best friends: Grisel and Nelly, and to my boyfriend Osvaldo. Your constant love, support and eternal encouragement gave me the strength and motivation to accomplish my goals. ii ACKNOWLEDGEMENTS I would like to thank my mentor Roger Sunahara for all his support and endless enthusiasm. His support and patience allowed me to become a great researcher. I would also like to thank all the members of the Sunahara lab, past and present, especially Dr. Matthew Whorton and Dr. Adam Kuszak. Thank you for all your help and motivation especially when science was not on my side. Thank you for the great science but most importantly for the laughs; not matter how bad my day was they always found a way to make me laugh and I was always happy to be in lab even if I was not working. I would also like to thank Brian DeVree for all his help and endless conversations about GPCR theory and the implications of our research; a lot of those were documented here. I was extremely fortunate to have great collaborators that not only provided me with all the reagents I needed but also were key in making my project a success. -

Physiological and Pathophysiological Functions of the Ecto-Nucleotide Pyrophosphatase/Phosphodiesterase Family
CORE Metadata, citation and similar papers at core.ac.uk Provided by Elsevier - Publisher Connector Biochimica et Biophysica Acta 1638 (2003) 1–19 www.bba-direct.com Review Physiological and pathophysiological functions of the ecto-nucleotide pyrophosphatase/phosphodiesterase family James W. Godinga, Bert Grobbenb, Herman Slegersb,* a Department of Pathology and Immunology, Monash Medical School, Monash University, Prahran 3181, Victoria, Australia b Department of Biomedical Sciences, Cellular Biochemistry, University of Antwerp, Universitaire Instelling Antwerpen, Universiteitplein 1, B-2610 Antwerpen-Wilrijk, Belgium Received 6 February 2003; received in revised form 20 March 2003; accepted 3 April 2003 Abstract The ecto-nucleotide pyrophosphatase/phosphodiesterase (E-NPP) multigene family contains five members. NPP1-3 are type II transmembrane metalloenzymes characterized by a similar modular structure composed of a short intracellular domain, a single transmembrane domain and an extracellular domain containing a conserved catalytic site. The short intracellular domain of NPP1 has a basolateral membrane-targeting signal while NPP3 is targeted to the apical surface of polarized cells. NPP4-5 detected by database searches have a predicted type I membrane orientation but have not yet been functionally characterized. E-NPPs have been detected in almost all tissues often confined to specific substructures or cell types. In some cell types, NPP1 expression is constitutive or can be induced by TGF-h and glucocorticoids, but the signal transduction pathways that control expression are poorly documented. NPP1-3 have a broad substrate specificity which may reflect their role in a host of physiological and biochemical processes including bone mineralization, calcification of ligaments and joint capsules, modulation of purinergic receptor signalling, nucleotide recycling, and cell motility. -

A Plant Proton-Pumping Inorganic Pyrophosphatase Functionally Complements the Vacuolar Atpase Transport Activity and Confers Bafilomycin Resistance in Yeast Jose R
A plant proton-pumping inorganic pyrophosphatase functionally complements the vacuolar ATPase transport activity and confers bafilomycin resistance in yeast Jose R. Perez-Castiñeira, Agustín Hernández, Rocio Drake, Aurelio Serrano To cite this version: Jose R. Perez-Castiñeira, Agustín Hernández, Rocio Drake, Aurelio Serrano. A plant proton-pumping inorganic pyrophosphatase functionally complements the vacuolar ATPase transport activity and con- fers bafilomycin resistance in yeast. Biochemical Journal, Portland Press, 2011, 437 (2), pp.269-278. 10.1042/BJ20110447. hal-00605257 HAL Id: hal-00605257 https://hal.archives-ouvertes.fr/hal-00605257 Submitted on 1 Jul 2011 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Biochemical Journal Immediate Publication. Published on 26 May 2011 as manuscript BJ20110447 Title: A plant proton-pumping inorganic pyrophosphatase functionally complements the vacuolar ATPase transport activity and confers bafilomycin resistance in yeast Short title: Functional complementation of yeast vacuolar H+-ATPase by a H+-PPase Authors: José R. PÉREZ-CASTIÑEIRA, Agustín HERNÁNDEZ, Rocío DRAKE and Aurelio SERRANO1 Instituto de Bioquímica Vegetal y Fotosíntesis, Universidad de Sevilla-CSIC, Avda. Americo Vespucio, 49, 41092 Sevilla, Spain 1 To whom correspondence should be addressed (Phone: +-34-954489525. -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S. -

Protein T1 C1 Accession No. Description
Protein T1 C1 Accession No. Description SW:143B_HUMAN + + P31946 14-3-3 protein beta/alpha (protein kinase c inhibitor protein-1) (kcip-1) (protein 1054). 14-3-3 protein epsilon (mitochondrial import stimulation factor l subunit) (protein SW:143E_HUMAN + + P42655 P29360 Q63631 kinase c inhibitor protein-1) (kcip-1) (14-3-3e). SW:143S_HUMAN + - P31947 14-3-3 protein sigma (stratifin) (epithelial cell marker protein 1). SW:143T_HUMAN + - P27348 14-3-3 protein tau (14-3-3 protein theta) (14-3-3 protein t-cell) (hs1 protein). 14-3-3 protein zeta/delta (protein kinase c inhibitor protein-1) (kcip-1) (factor SW:143Z_HUMAN + + P29312 P29213 activating exoenzyme s) (fas). P01889 Q29638 Q29681 Q29854 Q29861 Q31613 hla class i histocompatibility antigen, b-7 alpha chain precursor (mhc class i antigen SW:1B07_HUMAN + - Q9GIX1 Q9TP95 b*7). hla class i histocompatibility antigen, b-14 alpha chain precursor (mhc class i antigen SW:1B14_HUMAN + - P30462 O02862 P30463 b*14). P30479 O19595 Q29848 hla class i histocompatibility antigen, b-41 alpha chain precursor (mhc class i antigen SW:1B41_HUMAN + - Q9MY79 Q9MY94 b*41) (bw-41). hla class i histocompatibility antigen, b-42 alpha chain precursor (mhc class i antigen SW:1B42_HUMAN + - P30480 P79555 b*42). P30488 O19615 O19624 O19641 O19783 O46702 hla class i histocompatibility antigen, b-50 alpha chain precursor (mhc class i antigen SW:1B50_HUMAN + - O78172 Q9TQG1 b*50) (bw-50) (b-21). hla class i histocompatibility antigen, b-54 alpha chain precursor (mhc class i antigen SW:1B54_HUMAN + - P30492 Q9TPQ9 b*54) (bw-54) (bw-22). P30495 O19758 P30496 hla class i histocompatibility antigen, b-56 alpha chain precursor (mhc class i antigen SW:1B56_HUMAN - + P79490 Q9GIM3 Q9GJ17 b*56) (bw-56) (bw-22). -

Biophysical and Computational Landscapes of Mycobacterial Pyrophosphatase: a Closer View of Drug Repurposing with Different Natural and Synthetic Compounds
Biophysical and Computational Landscapes of Mycobacterial Pyrophosphatase: A Closer View of Drug Repurposing With Different Natural and Synthetic Compounds. Shivangi Shivangi CSIR Institute of Genomics & Integrative Biology Laxman Meena ( [email protected] ) CSIR-Institute of Genomics and Integrative Biology https://orcid.org/0000-0002-1356-496X Research Article Keywords: Mycobacterium tuberculosis, ppa, GTPase inhibitor, GTP, ITC Posted Date: March 16th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-295339/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/33 Abstract Mycobacterial pyrophosphatase (Mt-ppa) play essential role in bacterial in vitro and in vivo survival. This family of proteins reacts on pyrophosphates to release orthophosphates and protect bacteria from pyrophosphates toxicity. Rv3628 encodes pyrophosphate protein which is a type I pyrophosphate protein. This protein is engaged in hydrolysis of tri and diphosphates (majorly GTP, ATP and GDP) and its catalysis is metal ion dependent. Mt-ppa was showed ecient interaction with GTP molecule, whose Kd was 37.9µM, ΔH was -11Kcal/mol and ΔG was -6.06Kcal/mol. The protein was interacted with ATP family genes that resemble that it is participator in conversion of diphosphates moiety to the residual monophosphates. W102G, V150G, F44G, I119G, L93F, F3G, F122G, I108G, L32G, M82G, Y17G, L59G, V5G, V26G, I7G, W140D, W140G, W140A, F80G, W140S, L49G, L56G, I9G, V60G, V19G, V92G, L28G, L61G, Y126E and F123G are the top 30 mutation hits and Y126G, Y42G, R30G, E8G, K16G are top mutational hits in active site of Mt-ppa. Mt-ppa is temperature and pH sensitive as increasing temperature and pH decreases protein stability. -

POLSKIE TOWARZYSTWO BIOCHEMICZNE Postępy Biochemii
POLSKIE TOWARZYSTWO BIOCHEMICZNE Postępy Biochemii http://rcin.org.pl WSKAZÓWKI DLA AUTORÓW Kwartalnik „Postępy Biochemii” publikuje artykuły monograficzne omawiające wąskie tematy, oraz artykuły przeglądowe referujące szersze zagadnienia z biochemii i nauk pokrewnych. Artykuły pierwszego typu winny w sposób syntetyczny omawiać wybrany temat na podstawie możliwie pełnego piśmiennictwa z kilku ostatnich lat, a artykuły drugiego typu na podstawie piśmiennictwa z ostatnich dwu lat. Objętość takich artykułów nie powinna przekraczać 25 stron maszynopisu (nie licząc ilustracji i piśmiennictwa). Kwartalnik publikuje także artykuły typu minireviews, do 10 stron maszynopisu, z dziedziny zainteresowań autora, opracowane na podstawie najnow szego piśmiennictwa, wystarczającego dla zilustrowania problemu. Ponadto kwartalnik publikuje krótkie noty, do 5 stron maszynopisu, informujące o nowych, interesujących osiągnięciach biochemii i nauk pokrewnych, oraz noty przybliżające historię badań w zakresie różnych dziedzin biochemii. Przekazanie artykułu do Redakcji jest równoznaczne z oświadczeniem, że nadesłana praca nie była i nie będzie publikowana w innym czasopiśmie, jeżeli zostanie ogłoszona w „Postępach Biochemii”. Autorzy artykułu odpowiadają za prawidłowość i ścisłość podanych informacji. Autorów obowiązuje korekta autorska. Koszty zmian tekstu w korekcie (poza poprawieniem błędów drukarskich) ponoszą autorzy. Artykuły honoruje się według obowiązujących stawek. Autorzy otrzymują bezpłatnie 25 odbitek swego artykułu; zamówienia na dodatkowe odbitki (płatne) należy zgłosić pisemnie odsyłając pracę po korekcie autorskiej. Redakcja prosi autorów o przestrzeganie następujących wskazówek: Forma maszynopisu: maszynopis pracy i wszelkie załączniki należy nadsyłać w dwu egzem plarzach. Maszynopis powinien być napisany jednostronnie, z podwójną interlinią, z marginesem ok. 4 cm po lewej i ok. 1 cm po prawej stronie; nie może zawierać więcej niż 60 znaków w jednym wierszu nie więcej niż 30 wierszy na stronie zgodnie z Normą Polską. -

Supplementary Information For
Supplementary Information for Mechanisms for achieving high speed and efficiency in biomolecular machines Jason A. Wagoner and Ken A. Dill1 1To whom correspondence should be addressed. E-mail: dilllaufercenter.org This PDF file includes: Supplementary text Fig. S1 Tables S1 to S4 References for SI reference citations Jason A. Wagoner and Ken A. Dill1 1 of 13 www.pnas.org/cgi/doi/10.1073/pnas.1812149116 Supporting Information Text 1. Flux for the two-state model The steady-state flux (number of full cycles per unit time) can be derived from J = PBfm − PArm, where the transition rates are labelled in Figure 1 of the main text, and PA, PB are steady state probabilities that can be solved using the 2 × 2 rate matrix. Here, we give a derivation of the steady state flux that can be used for a more general network of states and for other kinetic properties, like higher moments of flux. The evolution of probability density along the periodic two-state model is ∂P (A , t) j = P (B , t) f + P (B , t) r ∂t j−1 m j c −P (Aj , t) (1 − fc − rm) , [S1] where P (Aj , t) is the probability density of state Aj at time t. The steady state flux can be calculated from the generating functions X −ijk P˜A (k, t) = e P (Aj , t) , [S2] j and similarly for P˜B (k, t). The time evolution of the generating function is ∂P˜A (k, t) X = M P˜ (k, t) , [S3] ∂t An n n∈{A,B} where −ik 1 − fc − rm rc + fme M = ik [S4] fc + rme 1 − fm − rc, and we are using lettered rather than numbered indices (MAB is M12, etc.).